Asthma Clinical Trials
Pedro C. Avila
Clinical trials are the gold standard to prove efficacy of new therapies and management approaches for any disease. These trials take place after preclinical development of new drugs usually based on our understanding of disease pathogenesis. Drugs vary widely from low-molecular-weight molecules to large recombinant compounds such as monoclonal antibodies. Once a candidate drug is selected for development, it is submitted to several toxicology tests before the U.S. Food and Drug Administration (FDA) approves it for investigational studies in humans. Under FDA supervision, clinical development takes place with phases I, II, and III studies to learn the pharmacokinetics, pharmacodynamics, and clinical efficacy of the new drug. Several drugs have been developed for asthma, targeting airway inflammation and relaxation of airway smooth muscle. As our understanding of asthma pathogenesis evolves, new drugs are being developed to manage a disease that affects 20 million Americans and 300 million worldwide.
Development of a Drug
A drug undergoes several phases of development until it is marketed (Fig.22.1) (1). A drug is initially developed from the understanding of the essential biological processes in disease pathogenesis based on human studies and animal models of human disease. One approach to developing a drug is to model a critical step of a pathway in an in vitro system (e.g., biological response to a cytokine receptor) and test several compounds to identify which ones affect the pathway. Pharmaceutical industries have libraries with thousands of natural and synthetic chemicals, peptides, nucleic acids, and other organic molecules which can be screened in high throughput assay systems to test their biological activity. Another approach is to design new compounds based on crystallography three-dimensional structure and computer drug design. In this case, scientists use crystallography to discover the three-dimensional structure of the target molecules and how they spatially interact (e.g., ligand-receptor binding). Using this information, they use computer programs to design drugs, atom by atom, creating a three-dimensional molecule that can potentially disturb the spatial interaction between the target molecules. These designed drugs are then synthesized and tested in biological systems for their activities. A third approach is to use biotechnology to produce recombinant molecules that are agonists (e.g., a cytokine) or antagonists (e.g., monoclonal antibodies, soluble receptors) in the target pathway. In the case of monoclonal antibodies, they were initially produced in mouse cell systems, but the latest biotechnology allows humanization of these antibodies by changing murine IgG antibody protein sequences to human IgG sequences except for the principal amino acids responsible for binding to the target epitope. This humanization of the antibody minimizes immunogenicity while sparing specificity and biological activity. For example, omalizumab and mepolizumab were originally murine monoclonal antibodies—against IgE and interleukin 5, respectively—that were humanized by changing most of their amino acids to human IgG sequences (2,3). Besides antibodies, biotechnology has produced recombinant human molecules for asthma trials such as interleukin-4 soluble receptor (4), interferon gamma (5), and interleukin 12 (6). Still newer approaches are under development such as using human cell systems to produce human monoclonal antibodies, genetic alteration of animals to produce human molecules (e.g., making goats secrete human growth hormone in the milk), DNA vaccines, virus vectors for human gene therapy, and epigenetic tools (e.g., small interfering RNAs).
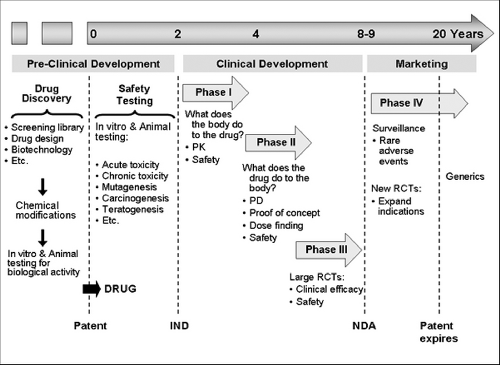 Figure 22.1 Phases of drug development. IND, Investigational New Drug approval; NDA, New Drug Application approval; PD, pharmacodynamics; PK, pharmacokinetics; RCTs, randomized clinical trials. |
A drug is usually developed together with several chemically similar counterparts. These similar compounds are tested in biological systems in vitro and in animal models of human disease for their biological activities to eventually identify a single or a few compound(s) for further development. These compounds may undergo chemical modifications to improve their eventual clinical application because a lot is known about organic chemical characteristics necessary for resistance to gastrointestinal digestion and successful oral absorption (bioavailability), to prolong half-life by affecting distribution and metabolism, and to avoid
toxicity. After in vitro testing, the drug is tested in animals for bioavailability, biological activity, specificity of action, and toxicity. After a lead compound is selected, the pharmaceutical company files for a patent to obtain exclusive rights to market it for 20 years. Next, the drug enters the pre-clinical phase of development to establish an extensive safety profile of the drug in standard animal and cell culture systems. This phase includes in vitro and animal experiments to assess dose range, lethal dose 50% (dose that kills 50% of the exposed animals) acute and chronic toxicity, teratogenesis, mutagenesis, carcinogenesis, effects on pregnancy, etc. During this phase the pharmaceutical company discusses with the FDA what safety data in animals will be required to start human studies. Once the pharmaceutical company and the FDA discuss the design of the first human study with the new drug, the company submits an Investigational New Drug (IND) application. Only after the FDA approves the IND can the first clinical trial begin, initiating the clinical phase of drug development.
toxicity. After in vitro testing, the drug is tested in animals for bioavailability, biological activity, specificity of action, and toxicity. After a lead compound is selected, the pharmaceutical company files for a patent to obtain exclusive rights to market it for 20 years. Next, the drug enters the pre-clinical phase of development to establish an extensive safety profile of the drug in standard animal and cell culture systems. This phase includes in vitro and animal experiments to assess dose range, lethal dose 50% (dose that kills 50% of the exposed animals) acute and chronic toxicity, teratogenesis, mutagenesis, carcinogenesis, effects on pregnancy, etc. During this phase the pharmaceutical company discusses with the FDA what safety data in animals will be required to start human studies. Once the pharmaceutical company and the FDA discuss the design of the first human study with the new drug, the company submits an Investigational New Drug (IND) application. Only after the FDA approves the IND can the first clinical trial begin, initiating the clinical phase of drug development.
The clinical development of a drug involves three phases of clinical studies before a drug is approved for marketing. All studies in this phase are designed by the pharmaceutical company with continuous discussions and oversight of the FDA. In Phase I studies, the drug is administered to few humans (e.g., n=10) for the first time after completion of extensive animal studies. Phase I helps us to understand drug pharmacokinetics (PK) and begin to assess safety in humans. In addition, biomarkers of drug activity can be measured, although this is a secondary objective. At the end of phase I studies, researchers have the first data on adverse effects in humans, determine the maximum tolerated dose, and define the drug’s PK characteristics such as time to peak serum level, bioavailability, half-life, metabolism, volume distribution, and route of elimination.
Based on the dose that worked in animal studies and on PK data in humans, Phase II studies are designed to assess pharmacodynamics (PD), that is, to determine whether the drug causes the expected biological effects for the targeted human disease. Two kinds of studies are usually conducted in this phase: proof-of-concept and dose-finding studies. In proof-of-concept studies, the biological effect of the drug on the disease of interest is assessed in small randomized clinical trials where usually maximal dose is administered to determine if the new drug really works. In asthma, a common proof-of-concept study is to assess the inhibitory effect of the drug on the early and late airway responses to inhaled allergen challenge. Often pharmaceutical companies conduct a couple of proof-of-concept studies that are considered “go” or “no-go” trials; that is, if the drug
shows no signs of efficacy in these trials, clinical development is stopped. In a dose-finding trial, a few hundred subjects are randomized to placebo or 2 or more different dose regimens in a double-blind fashion to determine what doses improve disease-related outcomes (e.g., forced expiratory volume in 1 second [FEV1] in asthma). Surrogate biological markers of efficacy are often used to enable these trials to be short and thus less costly. Such secondary outcomes may include assays in patients’ samples to determine whether the drug had the expected biological effects in the targeted pathway. For example, in trials of omalizumab, a neutralizing anti-IgE antibody, besides asthma clinical outcomes, serum-free IgE concentration was also measured as a surrogate marker to titrate dose to drive free IgE to undetectable levels. At the end of phase II trials, researchers know the dose that affects important disease physiological outcomes and have additional safety data in hundreds of individuals. This information is then used to plan and design phase III clinical trials to assess efficacy aiming at obtaining the necessary data on efficacy in clinically important outcomes and safety data to apply for FDA approval to market the drug.
shows no signs of efficacy in these trials, clinical development is stopped. In a dose-finding trial, a few hundred subjects are randomized to placebo or 2 or more different dose regimens in a double-blind fashion to determine what doses improve disease-related outcomes (e.g., forced expiratory volume in 1 second [FEV1] in asthma). Surrogate biological markers of efficacy are often used to enable these trials to be short and thus less costly. Such secondary outcomes may include assays in patients’ samples to determine whether the drug had the expected biological effects in the targeted pathway. For example, in trials of omalizumab, a neutralizing anti-IgE antibody, besides asthma clinical outcomes, serum-free IgE concentration was also measured as a surrogate marker to titrate dose to drive free IgE to undetectable levels. At the end of phase II trials, researchers know the dose that affects important disease physiological outcomes and have additional safety data in hundreds of individuals. This information is then used to plan and design phase III clinical trials to assess efficacy aiming at obtaining the necessary data on efficacy in clinically important outcomes and safety data to apply for FDA approval to market the drug.
Phase III studies are large double-blind, placebo-controlled, randomized clinical trials to determine whether the drug improves clinically relevant outcomes. For asthma some of the main outcomes to establish efficacy are airway obstruction, symptoms, quality of life, and frequency of exacerbations (see Table 22.1). When phase III studies are conducted, the pharmaceutical company and the FDA have a clear idea of what efficacy and safety data will be needed for drug approval. The FDA usually requires more than one phase III trial demonstrating efficacy. If phase III trials are successful, the pharmaceutical company submits to the FDA a new drug application to obtain approval for marketing. This application contains all data available on the drug since its preclinical development, and therefore contains several thousands of pages, including data on drug administration to 3,000 to 5,000 subjects. The FDA takes on average 6 months to approve or deny a new drug application (range: 3 months to years) and
may seek input from outside experts. Once approved, the pharmaceutical company can market the drug with exclusivity until the patent expires, at which point other companies can start producing and marketing the drug without having to pay a fee to the patent holder. After approval, new phase III studies can be undertaken to expand indications to different age groups (e.g., pediatrics) and new diseases, which can help extend duration of patents.
may seek input from outside experts. Once approved, the pharmaceutical company can market the drug with exclusivity until the patent expires, at which point other companies can start producing and marketing the drug without having to pay a fee to the patent holder. After approval, new phase III studies can be undertaken to expand indications to different age groups (e.g., pediatrics) and new diseases, which can help extend duration of patents.
Table 22.1 Outcomes in Asthma Clinical Trials | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
After FDA approval, phase IV studies are designed to monitor safety aiming at identifying severe and rare side effects such as those occurring at a rate of 1:10,000 or rarer. Examples of rare adverse events discovered in this phase include liver toxicity caused by telithromycin, cardiovascular events caused by rofecoxib, progressive multifocal leukoencephalopathy caused by JC virus in those receiving rituxan (anti-CD20), tendon rupture in patients taking quinolones, and possibly, increased risk of asthma-related death in patients taking long-acting bronchodilators, particularly in African Americans. Besides phase IV trials, these rare serious events can be captured through the FDA surveillance system for medications’ adverse events called MedWatch (http://www.fda.gov/medwatch/), which allows health care professionals to report adverse events directly to the FDA online. Rare serious adverse events proven to be related to a drug lead to “black box warnings” in the drug’s package insert, limitations of FDA-approved indications based on new risk:benefit ratio assessments, and even drug withdrawal from the market.
The costly development of new drugs is a risky business. It is estimated that the cost to bring a new drug to market is about $900 million. Many drugs fail during clinical development and only 30% of marketed drugs return the costs for their development. New drugs can fail even after marketing in phase IV studies because of rare life-threatening adverse events leading to restrictions in use or withdrawal from the market. Although patents protect marketing of new drugs for 20 years, it usually takes 8 to 10 years to obtain FDA approval to market a drug, leaving 10 or fewer years for the pharmaceutical company to profit from the drug. This profit not only should cover the expenses incurred to develop the drug itself, but also fund research and development of new drugs to keep the pharmaceutical company in business. Successful drugs can be highly profitable such as atorvastatin (Lipitor) with over $10 billion dollars in sales in 2004, or fluticasone-salmeterol inhaler (Advair) with almost $8 billion dollars in sales in 2008.
Outcomes in Asthma Trials
Outcomes measured in asthma trials have evolved as did our knowledge of asthma pathogenesis, clinical trials science, and biomedical research technology. In the early 1900s, pathological and clinical evidence already indicated that asthma pathogenesis involved bronchoconstriction, eosinophilic bronchitis, and exposure to external allergen triggers, which also caused hay fever. With the advent of pulmonary function testing in the 1940s to 1950s, it was noticed that patients with asthma had reversible airway obstruction and airway hyperresponsiveness. In the 1970s, it was documented that inhalation allergen challenges caused early and late airway bronchoconstriction associated with increased blood eosinophilia. In the 1980s, bronchoscopic biopsies of bronchial mucosa revealed chronic airway inflammation even in the mild cases, which is characterized mainly by eosinophilic bronchitis, and increase in CD4+ T cells. In the 1990s, remodeling was described, which entails alterations in the resident cells resulting from chronic inflammation driven by infiltrating leukocytes. Remodeling includes goblet-cell hyperplasia, smooth-muscle-cell hyperplasia, collagen deposition in the subepithelial reticular membrane, and increased vasculature, among other changes (7). Currently, research continues to focus on mechanisms of inflammation, particularly inflammatory mediators produced by resident cells, innate response, and interactions between resident cells and leukocytes.
The number and variety of clinical outcomes measured in asthma trials expanded based on our understanding of pathogenesis of airway disease as aforementioned. Initial trials measured spirometric outcomes such as FEV1 to assess changes in airway caliber. Later, portable peak expiratory flow (PEF) meters allowed patients to monitor airway flow at home. More recently, electronic portable devices can measure and record PEF, FEV1 and forced vital capacity (FVC) of 6 seconds, greatly expanding our ability to monitor variability in airway obstruction, a hallmark of asthma.
Airway hyperresponsiveness to nonspecific stimuli is also measured in asthma trials because it is an important feature of asthma (8,9) and because it correlates with airway inflammation. It is commonly measured as the provocative concentration of methacholine or histamine to cause a 20% decline in FEV1 (PC20). Airways of asthmatic subjects undergo excessive bronchoconstriction upon inhalation of methacholine or histamine, which acts directly on smooth muscles causing contraction. Airway hyperactivity is defined as a PC20 less than 8 mg/mL for direct agents (10). Inhaled corticosteroid therapy simultaneously improves both hyperresponsiveness and airway inflammation. Indirect agents are also used to assess PC20. They cause bronchoconstriction indirectly by stimulating mast cells to release bronchospastic mediators including histamine, cysteinyl leukotrienes, and prostaglandin D2. Examples of indirect agents to assess airway responsiveness include exercise, inhalation of adenosine, or inhalation of osmotic stimulants such as cold dry air, distilled water, hypertonic saline, or mannitol (11). PC20 using indirect agents can correlate more closely with airway
inflammation than PC20 using direct agents (e.g., methacholine and histamine). Airway hyperresponsiveness can also occur in medical conditions other than asthma, including allergic rhinitis without asthma, up to 6 weeks after respiratory virus infections, and in smokers with chronic obstructive pulmonary disease (10).
inflammation than PC20 using direct agents (e.g., methacholine and histamine). Airway hyperresponsiveness can also occur in medical conditions other than asthma, including allergic rhinitis without asthma, up to 6 weeks after respiratory virus infections, and in smokers with chronic obstructive pulmonary disease (10).
The realization that IgE-sensitization and inhalation of a relevant allergen caused reproducible early and late airway responses (EAR and LAR) led to the development of a widely used proof-of-concept asthma study model. In this model, subjects inhaled increasing amounts of allergen to which they reacted in allergy skin testing to determine the concentration of allergen that causes a 20% decline in FEV1. Then, the subject receives placebo and/or drug therapy for a period of time and returns for a repeat allergen challenge using the same allergen and dose as the initial challenge to determine whether the drug attenuated airway responses to the allergen. Because EAR and LAR are very reproducible, only 10 to 12 patients are needed per group to assess whether a drug attenuates any response by 30% or more. Almost all currently available asthma drugs attenuate EAR, LAR, or both (2,12–25), making this study model a common phase II trial to determine whether a new drug works for asthma (Fig. 22.2). Drugs that inhibit mast cell activation and bronchoconstriction should attenuate EAR, whereas drugs that inhibit delayed production of mediators or airway influx of leukocytes (e.g., eosinophils, dendritic cells, and lymphocytes) can inhibit LAR. This model of inhalation allergen challenge to induce 20% decline in FEV1 also increases airway hyperresponsiveness and sputum eosinophilia 24 hours after the challenge, allowing
researchers to assess the effects of new drugs in these outcomes as well. Presence of LAR seems to be driven by T cells because studies of peptide immunotherapy revealed late phase reactions to injections (26). BB Peptides are too small to cross-link IgE and stimulate mast cells, but they do bind to human leukocyte antigens and stimulate T cells. Lastly, EAR and LAR responses in the lower airways are not unique to asthmatic patients. They can also be present in nonasthmatic allergic rhinitic patients after rhinovirus colds (27) raising the hypothesis that a spectrum of disease progression in the lower airways may occur such as IgE-sensitization, bronchial hyperresponsiveness, bronchial EAR and LAR to allergen challenge, and finally full-blown asthma.
researchers to assess the effects of new drugs in these outcomes as well. Presence of LAR seems to be driven by T cells because studies of peptide immunotherapy revealed late phase reactions to injections (26). BB Peptides are too small to cross-link IgE and stimulate mast cells, but they do bind to human leukocyte antigens and stimulate T cells. Lastly, EAR and LAR responses in the lower airways are not unique to asthmatic patients. They can also be present in nonasthmatic allergic rhinitic patients after rhinovirus colds (27) raising the hypothesis that a spectrum of disease progression in the lower airways may occur such as IgE-sensitization, bronchial hyperresponsiveness, bronchial EAR and LAR to allergen challenge, and finally full-blown asthma.
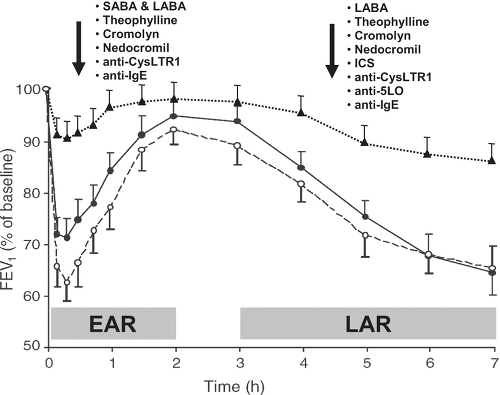 FIGURE 22.2 Inhalation allergen challenge causing biphasic airway response in asthmatic patients. Graph shows percentage changes in mean FEV1 and SEM from baseline to 7 hours post-challenge. Changes are reproducible between inhalation challenges performed 4 weeks apart (traced and continuous lines). Bronchoconstriction occurs within minutes and improves in 2 hours (early airway response—EAR) mostly as a consequence of smooth cell contraction. Then, 3-8 hours after challenge bronchoconstriction recurs as a consequence of increased influx of leukocytes, particularly eosinophils, TH2 lymphocytes and basophils (late airway response—LAR). Listed are medications that inhibit EAR and LAR (dotted line) when administered before the challenge. Anti-CysLTR1, antagonists of cysteinyl leukotriene receptor 1 (e.g. montelukast, zafirlukast); Anti-5LO, antagonist of 5-lipoxygenase (e.g. zileuton); Anti-IgE, antibody anti-IgE (e.g. omalizumab); ICS, Inhaled corticosteroids; LABA, long-acting beta-2 receptor agonist bronchodilators (e.g. salmeterol, formoterol); SABA, short-acting beta-2 receptor agonist bronchodilators (e.g. albuterol, terbutaline). |
Another common proof-of-concept study design used in phase II trials to assess efficacy of new drugs that aim at chronic asthma control rather than acute relief of bronchospasm is the corticosteroid withdrawal model. In this model, subjects with moderate-to-severe asthma enter a run-in period on inhaled plus or minus oral corticosteroid therapy that is titrated to the minimum corticosteroid dose necessary to control symptoms, at which point subjects are randomized to placebo or the new drug as an add-on therapy. Then, after a period on corticosteroid therapy and study medication (either active drug or placebo), corticosteroid therapy is tapered to determine whether the new drug is more efficacious than placebo in maintaining asthma control. In this type of study, patients need to be monitored very closely and protocols for action plan need to be in place to rescue patients when their asthma deteriorates. The corticosteroid withdrawal model is sometimes mistakenly called asthma exacerbation model. This is incorrect because exacerbations are caused by common colds in up to 80% of the episodes (28,29). Exacerbations occur when virus-induced inflammation superimposes to chronic allergen-driven inflammation. Studies evaluating exacerbations recruit patients who have exacerbated in the previous year and follow them for a long period (e.g., 12 or more months) to capture new episodes of acute asthma deterioration requiring a short course of systemic corticosteroid therapy. The corticosteroid withdrawal model is a model of loss of asthma control of chronic allergen-driven inflammation for lack of sufficient controller therapy.
The recognition that asthma is a chronic inflammatory airway condition led to implementation of measurements of inflammation in clinical trials (see Table 22.1). Blood eosinophilia was an initial marker. Bronchoalveolar lavage and bronchial mucosal biopsy reliably assess luminal and tissue inflammatory infiltrates, but necessitate bronchoscopy precluding their use in large clinical trials. In the 1990s, sputum induction using hypertonic saline solution started to be used in asthma studies as a noninvasive technique to assess airway inflammation. Sputum eosinophilia (more than 2% of nonsquamous cells) is characteristic of asthma, increases after allergen challenge (30), and decreases with therapy, including systemic (31) or inhaled corticosteroids (32), leukotriene antagonists (33), and anti-IgE (omalizumab) (34). Mast cell stabilizers (cromolyn and nedocromil) improve symptoms of asthma and airway function, but have mild and inconsistent anti-inflammatory effects as measured by eosinophil count or products in airways and blood (35–38
 Immunology of IgE-mediated and Other Hypersensitivity Responses
Immunology of IgE-mediated and Other Hypersensitivity Responses
 Physiologic and Biologic Evaluation of Allergic Lung Diseases
Physiologic and Biologic Evaluation of Allergic Lung Diseases
 Erythema Multiforme, Stevens-Johnson Syndrome, and Toxic Epidermal Necrolysis
Erythema Multiforme, Stevens-Johnson Syndrome, and Toxic Epidermal Necrolysis
 Management of Acute Severe Asthma
Management of Acute Severe Asthma
 Nasal Polyposis, Sinusitis, and Nonallergic Rhinitis
Nasal Polyposis, Sinusitis, and Nonallergic Rhinitis
 Eosinophilic Esophagitis
Eosinophilic Esophagitis




Related posts:
Immunology of IgE-mediated and Other Hypersensitivity Responses
Physiologic and Biologic Evaluation of Allergic Lung Diseases
Erythema Multiforme, Stevens-Johnson Syndrome, and Toxic Epidermal Necrolysis
Management of Acute Severe Asthma
Nasal Polyposis, Sinusitis, and Nonallergic Rhinitis
Eosinophilic Esophagitis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
