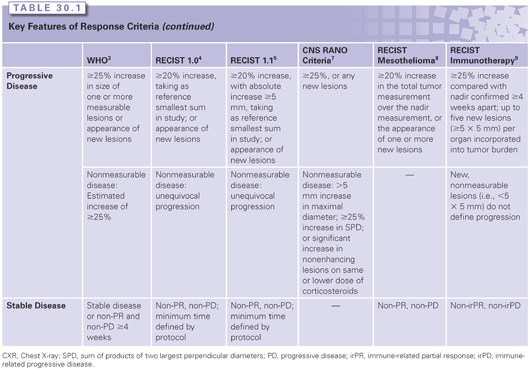
RECIST 1.1
The RECIST 1.0 guidelines were updated as RECIST 1.1 in 2009, with a number of differences between the two response criteria highlighted. RECIST 1.1 preserves the same categories of response found in RECIST 1.0:
■ Complete response: Complete disappearance of all disease
■ Partial response: ≥30% reduction in the sum of the longest diameter of target lesions
■ Stable disease: Change not meeting criteria for response or progression
■ Progression: ≥20% increase in the sum of the longest diameter of target lesions
However, a decade of experience with RECIST identified several problems with the criteria, some of which could be corrected. In RECIST 1.0, minimum size varied between 1 and 2 cm depending on technique; in RECIST 1.1, a 1-cm lesion is the minimum measurable. In RECIST 1.0, 10 lesions were to be measured, 5 per organ; RECIST 1.1 reduced that to 5 lesions, 2 per organ. Response criteria in RECIST 1.0 did not address lymph nodes; in RECIST 1.1, lymph nodes decreasing to <1 cm in their short axis could constitute a complete response. Disease progression in nontarget disease was further defined to indicate that in addition to a 20% increase in target lesions over the smallest sum on study, there must be an absolute increase of 5 mm, and that an increase of a single nontarget lesion should not trump an overall disease status assessment based on target lesions.
Variations of the RECIST Criteria
The RECIST criteria have been widely used for standardizing the reporting of clinical trial results and have improved reproducibility. However, the increasing precision and codification of RECIST has led to recognition of its limitations. For example, there are unique challenges in central nervous system (CNS) disease, relating response to tumor size measurements based on contrast enhancement. Pseudoprogression refers to an increase in contrast enhancement due to a transient increase in vascular permeability after irradiation, whereas pseudoresponse is a decrease in contrast enhancement that may occur due to a reduction in vascular permeability following corticosteroids or an antiangiogenic agent such as bevacizumab.10–12 The McDonald criteria, traditionally used in determining glioma response based on two-dimensional measurements, have been recently updated as part of the Response Assessment in Neuro-Oncology (RANO) response criteria and extended to include a response assessment for metastatic CNS disease.7,13
Other examples where RECIST is limited include mesothelioma, gastrointestinal stromal tumors (GIST), hepatocellular cancers, among others. The pleural disease of mesothelioma increases in depth while following the pleural surface. GIST tumors may remain unchanged in size after treatment, whereas the center of the tumor mass undergoes necrosis, and progression may occur in the remaining rim.14 Hepatocellular cancers are often treated with local–regional therapy in which the goal is tumor necrosis and treatment failure occurs in surviving viable tumor.15 Different strategies have emerged to quantify these diseases, including modifications of RECIST, quantifying positron-emission tomography (PET) imaging, and biomarker criteria, as will be discussed. The RECIST adaptation for mesothelioma, growing along the pleural surface, is to measure the diameter perpendicular to the chest wall or mediastinum, and to measure at three levels.8 The adaptation for hepatocellular cancer following local therapy is measurement of the longest diameter of the tumor that shows enhancement on the arterial phase of the scan, bypassing the dense, homogeneous Lipiodol-containing necrotic area.15
Investigators have also observed that following immunotherapy, tumor lesions may increase in size due to the increased infiltration of T cells, even meeting criteria for RECIST-defined progressive disease (PD). Previously radiographically undetectable lesions may appear. Departing from conventional RECIST, which defines any new lesion as PD, the immune response criteria allow the appearance of new lesions, adding them to the total tumor burden.9 An increase in total tumor burden of >25% relative to baseline or nadir is required to define PD.
International Working Group Criteria for Lymphoma
Revised guidelines for lymphoma assessment were promulgated by the International Working Group (IWG) in 2007.16 These guidelines incorporated 18F-fluorodeoxyglucose (FDG)-PET assessments in metabolically active lymphomas.16 Although a CR requires the complete disappearance of detectable disease, a posttreatment residual mass is permitted if it is negative on FDG-PET and was positive at baseline. For lymphomas that are not consistently FDG avid, or if FDG avidity is unknown, a CR requires that nodes >1.5 cm before therapy regress to <1.5 cm, and nodes that were 1.1 to 1.5 cm in long axis and >1.0 cm in the short axis shrink to ≤1.0 cm in short axis. The definition of PR resembles the WHO criteria, in that a ≥50% decrease in the sum of the product of the diameters in up to six nodal masses or in hepatic or splenic nodules must be documented. Although RECIST 1.1 now includes lymph node assessment, the IWG criteria remain the assessment method typically used in lymphoma clinical trials.
The previous examples represent attempts to more accurately measure tumor burden. Evolving imaging technology enabling volumetric measurements of tumor masses may eventually resolve some of these problems, but effective therapeutic agents are required to enable validation and utilization of response assessment tools. The lack of an agent that can mediate substantial tumor shrinkage underlies the concept of clinical benefit response (CBR) as an endpoint in pancreatic cancer. Clinical benefit was defined as a combination of improvement in pain, performance status, and weight; the assessment of CBR supported the U.S. Food and Drug Administration (FDA) approval of gemcitabine in pancreatic cancer.17,18 Better therapies for pancreatic cancer that result in tumor shrinkage or eradication should include and then eclipse clinical benefit.
Response criteria may be specific to a particular disease or clinical setting. Some diseases by their nature require specific strategies for response assessment.
Severity-Weighted Assessment Tool Score in Cutaneous T-Cell Lymphoma
Cutaneous T-cell lymphoma (CTCL) is a disease that can involve the entire epidermis, or comprise individual skin lesions varying widely in severity rather than size. The severity-weighted assessment tool (SWAT) assigns a factor for skin lesion severity—patch, plaque, or tumor—multiplies this factor by the percent of skin involved with each lesion type and then adds these together. This complex system formed the basis of the FDA approval of vorinostat for CTCL.19
Pathologic Complete Response in Breast Cancer
One unique response endpoint is the assessment of breast cancer treated in the neoadjuvant setting. The purpose of neoadjuvant therapy is to improve survival, render locally advanced cancer amenable to surgery, or to aid in breast conservation. In that setting, the absence of cancer cells in resected breast tissue has been used to define a pathologic complete response (pCR). The rate of pCR has been proposed as a surrogate endpoint for event-free survival (EFS) or overall survival (OS) to support approval of new agents or combinations of agents tested in clinical trials.20 In a pooled analysis of 11,955 patients enrolled on 12 neoadjuvant trials, individual patients with pCR had improved EFS and OS.21 However, at the trial level, pCR rates did not correlate with EFS or OS, a problem likely due to heterogeneity of breast cancer subtypes among the trials. Despite this, pCR rates were recently used to support the approval of pertuzumab and trastuzumab in the neoadjuvant setting.21,22
Computed Tomography-Based Tumor Density
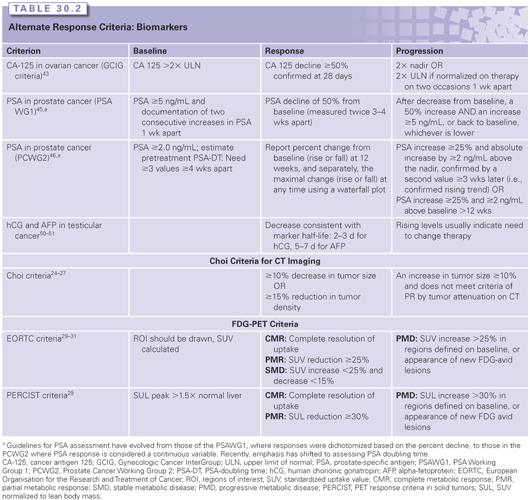
One approach, often called the Choi criteria, advocates assessing tumor response in GIST, renal cell cancer, or hepatocellular cancer based on density on computed tomography (CT) scans (Table 30.2). This variation was prompted by the evident response to treatment with imatinib but with minimal tumor shrinkage.23 The Choi criteria are still considered exploratory in GIST,24,25 and it is too soon to know of benefits in other histologies.26,27 Further study should determine its utility, although it will likely be confined to specific tumor types with specific drugs.
FDG-PET
Although widely used in clinical practice, FDG-PET has become part of standardized response criteria for clinical trials only in lymphoma (see Table 30.2). In solid tumors, FDG-PET can aid in the detection of new or recurrent sites of disease, and can be used as an adjunct during assessments for disease progression when using RECIST criteria.5 Although FDG uptake is a powerful diagnostic tool and its uptake reflects a tumor’s metabolic activity, it has some limitations: Some tumors have variable FDG avidity; differences can occur due to variations in patient activity, carbohydrate intake, blood glucose, and timing; and there are several benign sources of uptake, including inflammatory and postsurgical sites. Multiple methods of quantitating FDG-PET and assessing response have been proposed, but to date there is no consensus, particularly regarding the definition of a metabolic response.28–33
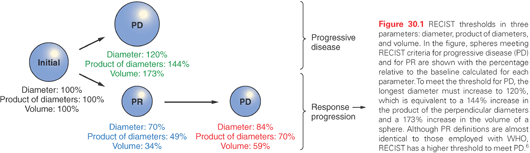
The two most widely used response criteria—the European Organisation for the Research and Treatment of Cancer (EORTC) criteria and PET Response Criteria in Solid Tumors (PERCIST) (see Table 30.2)—have been evaluated in specific disease types, but unifying FDG-PET response criteria remains a challenge in anticancer drug development.28,30 We would note that, as shown in Figure 30.1, a 30% reduction in the diameter of a sphere—the magnitude of change required to score a response according to RECIST—represents a 65% decrease in volume. If an standardized uptake value (SUV) decrease is directly equated to a volume decrease, a reduction of 25% translates to a 10% reduction in diameter, a value that likely constitutes an insufficient response.
Serum Biomarkers of Response
The ideal response assessment method is an assay that could measure tumor quantity by a simple blood test (see Table 30.2). Circulating protein biomarkers have been identified and studied for several decades for screening, early detection of recurrent disease, determining prognosis, selecting therapy, and monitoring response to therapy. These serum tumor markers are to be distinguished from the assays determining the presence of an overexpressed or mutated molecular target. With the successful launch of therapies against such molecular targets, there has been increased interest in the assays needed to select therapy for individual patients (predictive biomarkers). The analytical and clinical validation of such assays, along with determination of their clinical utility, has created a new regulatory paradigm known as companion diagnostics.34,35 This investment in the development of predictive markers for companion diagnostics has reduced the focus on protein biomarkers of treatment response relative to older literature.
As a result, there are few clinically validated biomarkers of response.36 In addition to issues regarding sensitivity and specificity, their use and development has also been hindered by the often limited efficacy of therapies; response biomarkers are of little value without highly effective primary and salvage therapies. For example, a recent clinical trial indicates that in asymptomatic patients with ovarian cancer whose only evidence of disease progression is an isolated rising CA-125, nothing is gained by instituting treatment before there is other evidence of progression.37,38
■ Cancer Antigen 125 (CA-125): Despite recognized limitations, CA-125 is widely used. For example, the Gynecologic Cancer InterGroup (GCIG) criteria have evolved to help determine whether a patient’s tumor has responded to therapy.39–41 Response is defined as a 50% decline from an elevated baseline value, whereas progression is defined as a doubling over the nadir or the upper limit of normal.42 In clinical practice, CA-125 levels are followed as part of standard management, but making clinical decisions on marker changes alone is not recommended.43
■ Prostate-Specific Antigen (PSA): Similar issues have confronted investigators caring for patients with prostate cancer. The PSA Working Group 1 (PCWG1) guidelines, first published in 1999, established PSA criteria, particularly for use in patients with disease that was difficult to quantify.44 There followed a second working group (PCWG2) that recommended plotting the percent PSA change for each patient in a waterfall plot so as to avoid creating a dichotomous variable from the changes in PSA.45 PCWG2 also recommended keeping patients on trial until evidence of a change in clinical status—either symptomatic or radiographic progression. The latter addressed concerns with patients in whom PSA changes did not reflect clinical status, particularly those with transient increases in the first 12 weeks of a new therapy.
■ Human Chorionic Gonadotropin (hCG) and alpha fetoprotein (AFP): Because testicular cancer is a highly curable disease with validated biomarkers, outcome assessment has focused on the rapid detection of patients whose tumors have a poor response to therapy. Because both markers have relatively short half-lives—2 to 3 days for hCG and 5 to 7 days for serum AFP—the rate of decline can be determined. Various methods have demonstrated that a rapid decline or early normalization of marker levels is indicative of a good outcome, without any one method achieving widespread acceptance.46–48 Nonetheless, the 2010 American Society of Clinical Oncology (ASCO) guidelines on serum tumor markers concluded there was still insufficient evidence to recommend changing therapy solely on the basis of a slow marker decline.49 Rising levels after two cycles of therapy (outside the first week of treatment when rises can be due to tumor lysis) can be considered an indication to change the treatment plan.49,50
Circulating Tumor Cells and Circulating Tumor DNA
Two response endpoints under recent investigation show a potential to detect the impact of therapy. One is the measurement of circulating tumor cells (CTC) in the bloodstream, enriched by one or more capture strategies, including one that has received FDA approval.51 The number of CTCs in the blood has been shown to be prognostic, with higher levels conferring a poor prognosis, and to correlate with a response to therapy. A second approach is the determination of levels of circulating tumor DNA (ctDNA) in the blood. This is detected by quantitating the number of DNA molecules carrying a given mutation or gene rearrangement in the blood, typically detected through targeted sequencing of common mutations, or of a previously identified mutation signature or gene rearrangement. The amount of ctDNA appears to correlate with tumor burden, increases with stage, and in one study, was deemed more sensitive than CTC detection.52–54 Whether these tests will ultimately prove to be more sensitive and accurate than the serum biomarkers discussed previously remains to be determined. Because targeted sequencing can be very sensitive, one concern is that false-positive ctDNA detection may occur after treatment, or intermittently in the setting of enlarging tumor masses. At the least, detection of CTCs and ctDNA is advancing our understanding of cancer biology, as studies reveal evidence of metastatic heterogeneity, clonal heterogeneity, and emergence of resistance mutations in clinical samples.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


