Arteriovenous Malformations and Hemangiomas and Cerebral Cavernous Malformations
Kevin J. Whitehead
Douglas A. Marchuk
Mark L. Kahn
The cardiovascular system is organized into a hierarchical, branching arrangement by which blood is distributed to, and collected from, organs through arteries, arterioles, capillaries, venules, and veins.1 This arrangement is not merely an elegant example of biologic patterning; it is also essential for physiologic control of blood flow throughout the body. Loss of the normal vascular architecture may result in localized or generalized vascular malformations in humans.2 Vascular malformations are descriptively categorized on the basis of the type of vessel that appears to be involved, for example, arterial, venous, or capillary. Most common vascular malformations, for example, arteriovenous malformations (AVMs) in which vascular connections link arteries and veins without an intervening capillary bed, arise spontaneously in affected individuals, but others arise in a heritable pattern that indicates contribution by germline mutations (e.g., cerebral cavernous malformation [CCM] and hereditary hemorrhagic telangiectasia [HHT], discussed further below).
Although many vascular malformations are not symptomatic, vascular malformations constitute an important component of pediatric and adult vascular disease. These vascular structures are fragile and frequently cause clinical disease due to hemorrhage in vulnerable sites such as the central nervous system (CNS) or the gastrointestinal (GI) tract. AVMs also come to clinical attention due to the cardiovascular effects of blood shunting such as high-output congestive heart failure. Since vascular malformations frequently arise in the skin, they are also an important component of dermatologic disease. The treatment of vascular malformations has improved in the past two decades but remains restricted to surgical and interventional vascular approaches to either remove them or block blood flow through them.
The lack of molecular pharmacologic therapies for vascular malformations reflects the fact that until very recently the pathogenesis of such lesions was virtually unknown. However, in the past decade, genetic studies of inherited human vascular diseases have intersected with developmental studies using model vertebrate organisms such as fish and mice to provide new molecular insights into the molecular and genetic basis for vascular malformations. These studies have revealed that many human vascular malformations arise because of loss of regulatory signaling pathways that participate in the development of the blood and lymphatic vascular systems. They have also produced useful animal models of human vascular malformations. Thus, there is hope that the new knowledge and biologic tools will enable the development of pharmacologic approaches to treat human vascular malformations. This chapter focuses on two common inherited vascular malformations, CCM and HHT (FIGURE 71.1).
CEREBRAL CAVERNOUS MALFORMATIONS
Clinical Presentation and Treatment
CCMs are thin-walled, dilated vascular malformations that present primarily in the CNS in early to mid adulthood.3,4 The prevalence of CCM in the general population is relatively high, 0.1% to 0.5%, making this malformation a relatively common condition. While the clinical symptoms associated with CCMs vary with the size and location of the malformation, seizures, headache, and stroke with associated neurologic deficits are the most common. CCMs are diagnosed by MRI, and more sophisticated imaging technologies have revealed that a majority of these lesions are clinically silent. Although the number of longitudinal studies is limited, existing studies indicate that in the case of familial CCM, both the number and size of CCMs increase over time.5 Presently, the treatment of CCMs is neurosurgical resection of symptomatic lesions. Histologic analysis of resected CCMs reveals vascular lesions with a thin endothelial lining that by transmission electron microscopy lack the typical number and quality of tight junctions between endothelial cells,6 an ultrastructural feature that has been valuable for comparison to animal models.
Genetics of CCM
Approximately one third of CCMs arise in patients with a family history of this disease.4 Familial CCMs are transmitted in an autosomal dominant pattern and are both more numerous and symptomatic than nonfamilial CCMs. Positional cloning studies have identified germline heterozygous loss-of-function mutations in three genes: KRIT1 (aka CCM1),7,8,9 CCM2,10,11 and PDCD10 (aka CCM3)12 that encode intracellular adaptor proteins without enzymatic activity. The fact that CCMs arise in older individuals who are heterozygous for a loss of function allele has led to the hypothesis that these lesions are created by a “second hit” that confers a complete loss of function in the causal cell type. Surgically resected lesions from individuals harboring germline CCM mutations have revealed second site mutations in the remaining wild-type copy of the relevant CCM gene, lending support to the two-hit mutation mechanism for CCM pathogenesis.13,14,15 Recent studies of mouse models also
support the two-hit mechanism and implicate the endothelial cell as the causal cell type.
support the two-hit mechanism and implicate the endothelial cell as the causal cell type.
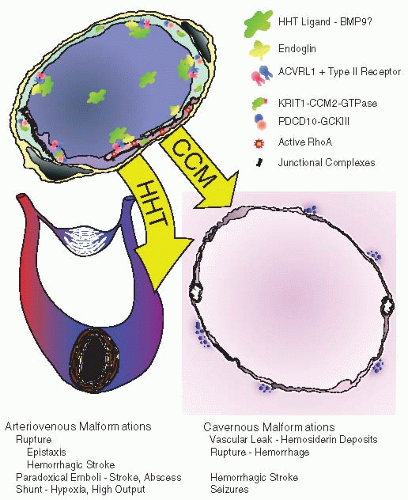 FIGURE 71.1 Pathogenesis of HHT and CCM vascular malformations. The endothelial cell signaling pathways that are affected in the pathogenesis of HHT and CCM are shown above. The resulting vascular malformations and their clinical manifestations are shown below. |
Biology of the CCM Signaling Pathway
Genetic studies of mice and zebrafish have revealed that the CCM proteins function in a novel signaling pathway that is required for vertebrate cardiovascular development.16,17,18,19,20,21,22,23,24,25,26 Despite the vascular phenotypes exhibited by deficient animals and humans, expression studies in mice have revealed that CCM1, CCM2, and CCM3 are broadly expressed genes and are particularly highly expressed in the CNS, a finding that has led to the hypothesis that this pathway regulates vascular function indirectly through signaling in neural cell types. Mice constitutively deficient in the CCM proteins die soon after the heart and vessels are formed due to defects in the formation of early vascular structures such as the branchial arch arteries that connect the heart to the aorta in the early embryo, so that these animals do not have circulating blood. Deficient zebrafish embryos exhibit similar branchial arch arterial defects, as well as a “dilated heart” phenotype conferred by deficiency of the type I transmembrane protein heart of glass (HEG).23 Finally, biochemical studies have established that HEG, CCM1, CCM2, and CCM3 form a molecular signaling complex in which the HEG cytoplasmic tail binds CCM1, the CCM2 phosphotyrosine binding domain binds CCM1, and CCM3 binds CCM2 through an as yet unidentified mechanism.23,27,28,29
Electron microscopy of human CCMs, as well as studies in cultured endothelial cells and deficient mice and zebrafish suggest that one important endpoint of CCM signaling is positive regulation of endothelial cell-cell junctions.6,21,23,25,30,31 However, precisely how CCM signaling regulates endothelial junctions is not completely understood. Two effectors of CCM signaling are RAP1 and RHO, proteins known to regulate cell junctions. RAP1 binds to the CCM1 FERM (4.1 protein, Ezrin, Radixin, and Moesin) domain and it has been proposed that CCM signaling is linked to RAP1 activity and localization in endothelial cells.30,32 Several groups have demonstrated that loss of CCM proteins results in elevated levels of RHO-GTP and loosening of cellular junctions in cultured endothelial cells.21,25,31,33,34 Thus, negative regulation of RHO may be a mechanism by which the CCM pathway regulates cell junctions. Biochemical studies and genetic studies in zebrafish have identified the GCK-III family of serine/threonine kinases as potential CCM3 binding and signaling partners in vivo that may regulate RHO and endothelial cell junctions.19,25 In addition to regulating cell junctions, the CCM pathway may also maintain endothelial cell polarity and the formation of endothelial tubes.35 Thus, the available evidence strongly supports a role for CCM signaling in positively regulating endothelial cell junctions and hint at other roles that may be connected to vessel formation and function.
CCM Pathogenesis: Insights from Animal Models
Sequence conservation and observations that zebrafish and mice lacking CCM proteins exhibit similar vascular phenotypes have suggested that the function of this pathway might be sufficiently conserved to permit in-depth investigation of human disease using genetically modified animals. Indeed, genetic studies using animal models have played a pivotal role in elucidating both the function of the CCM signaling proteins in vivo and the pathogenesis of human CCMs. Analysis of deficient zebrafish and mice have revealed that CCM signaling is required in vertebrates for early cardiovascular development,16,17,23,25 a result that suggested a direct role in cardiovascular cell types such as endothelial cells. However, none of the CCM disease genes are highly or specifically expressed in cardiovascular cell types. Therefore, an early question was whether CCM proteins are required in vascular cell types such as endothelial cells or pericytes, or whether vascular CCMs arise due to loss of CCM signaling in adjacent tissues such as neural cells. Recent studies of mice in which CCM2 and CCM3 were conditionally deleted in neural, endothelial, and smooth muscle cells revealed that the defects in cardiovascular development observed in globally deficient animals arose because endothelial cells require these proteins.21,24 These studies, as well as genetic studies of endothelial cells isolated from human CCMs, strongly suggest that human CCMs arise due to loss of this pathway in endothelial cells. The recent generation of mice carrying conditional CCM alleles has allowed this hypothesis to be directly tested using transgenic animals in which Cre recombinase can be inducibly expressed in endothelial cells and other cell types. However, results of these studies are controversial. It has recently been reported that deletion of CCM2 in endothelial cells after birth does indeed result in the formation of CCMs in the brain and retina,22 providing strong evidence that human CCMs arise from loss of CCM function in endothelial
cells. However, deletion of CCM3 in neuroglia has also been reported to result in the formation of CCM in mice.36 At this time, the weight of evidence favors an endothelial cell mechanism, but further studies addressing the cell specificity of CCM protein deletion in these studies will be required to definitively answer this question.
cells. However, deletion of CCM3 in neuroglia has also been reported to result in the formation of CCM in mice.36 At this time, the weight of evidence favors an endothelial cell mechanism, but further studies addressing the cell specificity of CCM protein deletion in these studies will be required to definitively answer this question.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


