Chapter 23 Approach to the diagnosis and classification of blood diseases
Initial screening tests
Although the range of haematological tests available to support clinical and public health services is broad, it is often the simplest investigations that are most useful in indicating the diagnosis. Even poorly-resourced laboratories are usually able to provide an initial panel of tests such as haemoglobin concentration (Hb), white blood cell count (WBC) and platelet count (Chapter 26) and examination of a peripheral blood film for a differential leucocyte count (Chapter 3) and cellular morphology (Chapter 5). These screening tests will often enable the underlying pathological processes to be suspected promptly and point to a few key diagnostic tests. The investigation of specific haematological problems is covered in detail in Chapters 9 (iron deficiency anaemia), 10 (megaloblastic anaemia), 11, 12 and 13 (haemolytic anaemias), 14 (haemoglobinopathies) and 18, 19 and 20 (coagulation disorders).
Interpretation of Screening Tests
Results of laboratory screening tests should always be interpreted with an understanding of the limitations of the tests and the physiological variations that occur with sex, age and conditions such as pregnancy and exercise. Physiological variations in cell counts are detailed in Chapter 2. Abnormalities of red cells, white cells or platelets may be quantitative (increased or reduced numbers) or qualitative (abnormal appearance and/or function).
Quantitative Abnormalities of Blood Cells
Erythrocytosis
Secondary polycythaemia can generally be excluded by the clinical history and examination, assessment of serum erythropoietin concentration and arterial oxygen saturation, haemoglobin electrophoresis plus oxygen dissociation curve and abdominal ultrasound examination. The presence of splenomegaly is suggestive of PV and this diagnosis can be confirmed by demonstrating the JAK2 V617F mutation, which is present in 95% of patients.2 Only if this mutation (or one of the much less common JAK2 exon 12 mutations) is not detected is the measurement of total red cell and plasma volume necessary (Chapter 17).
Leucocytosis
Neutrophilia
Neutrophils are commonly increased during pregnancy and in acute infections, inflammation, alcohol intoxication, corticosteroid therapy and acute blood loss or red cell destruction. Neutrophilia with the neutrophils showing heavy cytoplasmic granulation (‘toxic’ granulation) is a common finding in severe bacterial infections. In the absence of any underlying cause, a high neutrophil count with immature myeloid cells suggests chronic myelogenous leukaemia (CML); cytogenetic and molecular studies to look for t(9;22) and the BCR–ABL1 fusion gene are indicated (Chapter 8).
Lymphocytosis
Lymphocytosis is a feature of certain infections, particularly infections in children. It may be especially marked in pertussis, infectious mononucleosis, cytomegalovirus infection, infectious hepatitis, tuberculosis and brucellosis. Lymphocytosis is also a common transient reaction to severe physical stress. Elderly patients with lymphoproliferative disorders, including chronic lymphocytic leukaemia and lymphomas, often present with lymphadenopathy and a lymphocytosis. Morphology and immunophenotyping of the cells combined with histological examination of a bone marrow trephine biopsy specimen (and if necessary other tissue biopsy) are used to classify these disorders and to give an indication of management and prognosis.3 It is occasionally difficult to differentiate between a reactive and a neoplastic lymphocytosis. In this situation, immunophenotyping, to provide evidence of light chain restriction and polymerase chain reaction for immunoglobulin or T-cell receptor gene rearrangements, may indicate the presence of a monoclonal population of lymphocytes, thereby supporting a diagnosis of neoplastic, rather than reactive, lymphoproliferation. If lymph nodes are enlarged, a lymph node biopsy for histology and immunohistochemistry may be helpful in diagnosis.
Monocytosis
A slight to moderate monocytosis may be associated with some protozoal, rickettsial and bacterial infections including malaria, typhus and tuberculosis. High levels of monocytes (monocyte count >1 × 109/l) in an elderly patient suggest chronic myelomonocytic leukaemia or, sometimes, atypical chronic myeloid leukaemia. Because these conditions fall into the myelodysplastic/myeloproliferative neoplasm group of disorders,4 the diagnosis would be supported by finding splenomegaly, quantitative and qualitative abnormalities in other cell lines or a clonal cytogenetic abnormality.
Eosinophilia
Eosinophilia is typically associated with allergic disorders including drug sensitivity, skin diseases and parasitic infections. In most cases, the cause is indicated by the clinical history, which should include details of all medications and foreign travel, and by examination of the stool and urine for parasites, cysts and ova. A diagnosis of chronic eosinophilic leukaemia is made if there is dominant eosinophilia with an increase in blast cells in the blood or marrow, or cytogenetic or molecular evidence of an abnormal myeloid clone.5 If no other cause for eosinophilia is found it is important, because of the therapeutic implications (i.e. responsiveness to imatinib), to confirm or exclude a diagnosis of eosinophilic leukaemia related to rearrangement of PDGFRA or PDGFRB (see p. 559).6 The idiopathic hypereosinophilic syndrome is an unusual cause of eosinophilia in which release of the contents of eosinophil granules results in damage to the heart, lungs and other tissues. It is defined by the presence of a peripheral blood eosinophil count of 1.5 × 109/l or greater for at least 6 months with resultant tissue damage. This is a diagnosis of exclusion, made only when detailed investigations exclude other possible causes of eosinophilia including systemic mastocytosis, eosinophilic leukaemia and eosinophilia associated with a phenotypically aberrant T-cell population or a neoplastic clone of T cells.
Thrombocytosis
Thrombocytosis is often associated with infectious and inflammatory conditions such as osteomyelitis and rheumatoid arthritis. Haematological causes of thrombocytosis include chronic blood loss, red cell destruction, splenectomy and rebound following recovery from marrow suppression. Under these circumstances, a moderately increased platelet count (e.g. 400–800 × 109/l) does not usually have any pathological implications. Primary thrombocythaemia is usually the result of a myeloproliferative neoplasm; rarely, it is an inherited condition. When there is isolated persistent thrombocytosis in a myeloproliferative neoplasm the diagnosis is essential thrombocythaemia (as long as a BCR–ABL1 fusion gene has been excluded). Thrombotic or haemorrhagic complications can occur but often the diagnosis is an incidental one.7 A significant proportion of individuals with essential thrombocythaemia have the JAK2 V617F mutation, which is associated with an increased risk of thrombosis.8 The criteria for this diagnosis are discussed on p. 559.
Anaemia
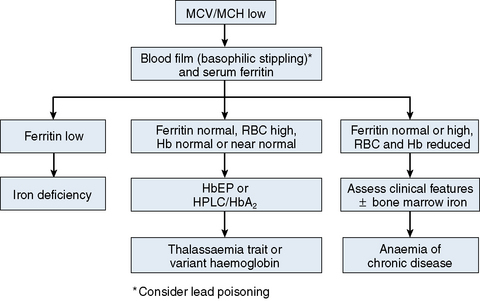
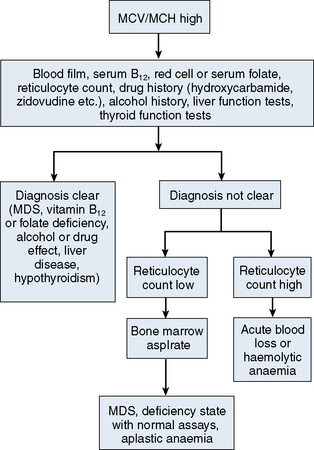
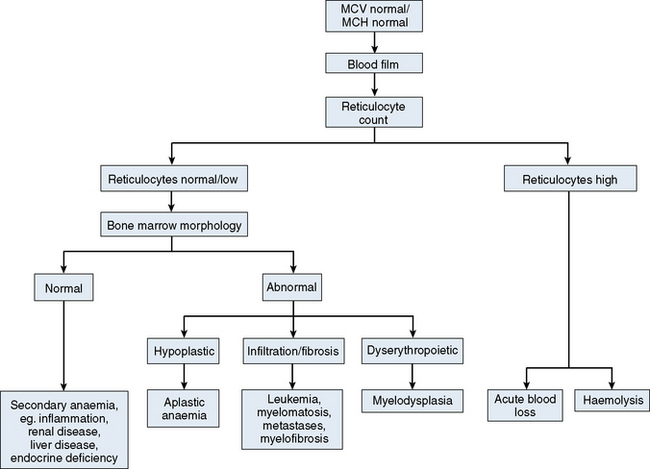
The mechanisms which result in anaemia are decreased production, reduced red cell lifespan, blood loss and splenic pooling. Anaemia is broadly divided into three types: microcytic (low MCV), macrocytic (high MCV) and normocytic (normal MCV). The choice of investigations is guided by the MCV and red cell morphology in addition to clinical features. Figures 23.1–23.3 are flow charts that provide an orderly sequence of investigations for the different types of anaemia on the basis of these indices. Examination of a blood film will usually suggest the quickest route to the diagnosis; confirmation may require the more specific tests, which are given in the text. The presence of basophilic stippling in a patient with microcytic red cells suggests thalassaemia trait or, much less often, lead poisoning. A dimorphic blood film is typical of congenital sideroblastic anaemia but is more often the result of iron deficiency responding to treatment. Pappenheimer bodies suggest that a microcytic anaemia is the result of sideroblastic erythropoiesis.
Microcytic Anaemia
The most common cause of anaemia worldwide is iron deficiency, which can be suspected from a low MCV (Fig. 23.1) and the presence of hypochromic, microcytic red cells. Laboratory confirmation of iron deficiency may be based on measurements of serum ferritin, serum iron plus either total iron-binding capacity or transferrin assay, red cell protoporphyrin and staining of bone marrow aspirates for iron (see Chapter 4). Assay of soluble transferrin receptors has good sensitivity and specificity for iron deficiency and may be useful in the presence of inflammation when interpretation of ferritin levels is difficult.9 A diagnosis of iron deficiency must be followed by a search for the cause. This should include specific questions relating to blood loss and dietary insufficiency and may require stool examination for parasites and occult blood, endoscopic examination of the gastrointestinal tract to exclude occult malignancy and tests for coeliac disease. The differential diagnosis of iron deficiency anaemia includes anaemia of chronic disease. Clinical and laboratory features of inflammation or chronic infection may suggest this diagnosis, which is confirmed by demonstration of normal or high serum ferritin and reduced serum iron, transferrin and iron-binding capacity.
The thalassaemias also cause microcytosis, but both α and β thalassaemia trait are usually associated with an increased red blood cell count (RBC) and a normal or near-normal Hb despite a considerable reduction of the MCV and MCH. In contrast, in iron deficiency the MCV and MCH do not fall until the Hb is significantly reduced. Further investigations, such as high-performance liquid chromatography (HPLC) or haemoglobin electrophoresis supplemented by measurement of Hb A2 and Hb F usually confirm the diagnosis of β thalassaemia trait. The diagnosis of α thalassaemia trait is more difficult; detection of infrequent Hb H inclusions is usually possible in α0 thalassaemia trait, but definitive diagnosis requires DNA analysis.10 A diagnosis of α0 thalassaemia heterozygosity can be clinically important for prediction of haemoglobin Bart’s hydrops fetalis since, if both parents have α0 thalassaemia, this very serious condition can occur in a fetus. DNA analysis is therefore indicated when a pregnant woman of appropriate ethnic origin has an MCH of <25 pg. Hb H inclusions may not be detected in α+ thalassaemia trait but this diagnosis is of less clinical importance and confirmation is thus not usually required.
Macrocytic Anaemia
A high MCV (Fig. 23.2) with oval macrocytes and hypersegmented neutrophils suggests folate or vitamin B12 deficiency and is an indication for assays of these vitamins (see Chapter 10); subsequent investigations could include malabsorption studies, tests for coeliac disease and tests for intrinsic factor and gastric parietal cell antibodies. In patients with these blood film findings and normal vitamin assays, haematinic deficiency is not completely excluded and further investigation is indicated. A Schilling test permits a definitive diagnosis of pernicious anaemia but currently this test is not available due to a lack of reagents. In the absence of intrinsic factor antibodies, the diagnosis of pernicious anaemia may be presumptive. Pernicious anaemia is commonly associated with autoimmune thyroid disease and other autoimmune disorders, such as diabetes mellitus. A high MCV may also be associated with alcohol excess and liver disease or use of drugs such as hydroxycarbamide or zidovudine. Macrocytosis resulting from chronic haemolysis is associated with increased numbers of immature red cells, which appear slightly larger and bluer than normal red cells (polychromatic macrocytes) on a Romanowsky-stained peripheral blood film. Supravital staining of blood films (see p. 33) or an automated reticulocyte count can be used to confirm reticulocytosis. Untreated anaemia associated with polychromasia is likely to indicate blood loss or haemolysis. The combination of red cell fragments, thrombocytopenia and polychromasia indicates a microangiopathic haemolytic anaemia and should trigger further tests such as a platelet count, coagulation studies, assessment of renal function and a search for infection or neoplastic disease. This further assessment is urgent because these may be features of thrombotic thrombocytopenic purpura, which requires immediate treatment by plasma exchange.
Normocytic Anaemia
Normochromic, normocytic anaemia (Fig. 23.3) is frequently the result of an underlying chronic, non-haematological disease. Investigations should include screening for renal insufficiency, subclinical infections, autoimmune diseases and neoplasia. In the presence of anaemia, a lack of polychromasia, confirmed by reticulocytopenia, points toward a primary failure of erythropoiesis or lack of compensatory increased red cell production in blood loss or haemolysis. Examination of the bone marrow may be helpful in demonstrating haematological causes for a normochromic, normocytic anaemia such as aplastic anaemia or MDS.11 Staining for iron may also show that there is a block in iron metabolism suggestive of anaemia associated with chronic inflammatory disease.




