Antithrombin and the Serpin Family
Susan C. Bock
ANTITHROMBIN III
Overview
Antithrombin III (ATIII) is a plasma proteinase inhibitor that inactivates thrombin and the enzymes responsible for the generation of thrombin. This combination of properties makes ATIII a very powerful and important endogenous anticoagulant molecule and explains why patients with even modest ATIII deficiencies experience clinical thrombosis. The anticoagulant activity of pharmaceutical heparins derive from their ability to potentiate the inhibitory activity of ATIII by mechanisms that are similar to physiologic activation of ATIII by vessel wall heparan sulfate proteoglycans (HSPGs). In addition to its wellknown anticoagulant activity, ATIII also has anti-inflammatory, antiproliferative, antiangiogenic, and antiviral properties that are related to its ability to block noncoagulant as well as coagulant functions of thrombin and to its ability to assume a variety of protein conformations.
Biochemistry
The plasma concentration of human ATIII1 is about 2.4 µM (i.e., 140 µg/mL).2 It circulates as two glycoforms: the predominant ATIII-α glycoform accounts for more than 90% of the ATIII in plasma, whereas the minor ATIII-β variant represents <10% of ATIII concentration.3 Both isoforms have an identical polypeptide backbone, for which the complete amino acid sequence is known.4,5 Disulfide bonds link cysteines 8 with 128, 21 with 95, and 247 with 430.5 The major α-ATIII isoform is modified with four identical sialylated, biantennary complex oligosaccharides on asparagines 96, 135, 155, and 192,6,7 and these account for 15% of the total mass of 58,000 Da. The minor β-ATIII isoform is not glycosylated on asparagine135, and some of its properties are distinct from those of the α-isoform, as discussed in the section on β-antithrombin. Both ATIII isoforms adopt several different protein conformations, which have distinct physical and functional properties. Crystal structures are available for several conformations and have considerably advanced the understanding of ATIII mechanism and regulation.8,9,10,11,12,13,14
The enzymes that are targets of ATIII inhibitory activity mediate blood coagulation, platelet activation, protease-activated receptor (PAR) signaling, and cell proliferation (see FIGURE 18.1). The name “antithrombin” is somewhat misleading because it suggests that ATIII is primarily an inhibitor of thrombin, when in fact, it has broad inhibitory activity for many different factors in the coagulation cascade. ATIII serves as an important regulator of hemostasis and thrombosis at several levels by blocking: (a) thrombin-mediated fibrin clot formation, (b) common pathway factor Xa (FXa)-mediated thrombin generation, and (c) coagulation factors that are higher up in the intrinsic and extrinsic pathways (factors IXa, XIa, XIIa [and its fragments], and plasma kallikrein [high molecular weight kininogen] and factor VIIa-tissue factor [TF]). ATIII inhibition of thrombin also serves to regulate its noncoagulant functions that mediate platelet activation and vascular cell signaling and proliferation (FIGURE 18.1).
Serpin Inhibition Mechanism
ATIII exhibits sequence, structural, and functional homology to members of the serpin (serine proteinase inhibitor) gene family.5,15 Many different serpins are present in blood and contribute to the regulation of the coagulation (ATIII), vascular remodeling (heparin cofactor II [HcII]), complement (C1 inhibitor), fibrinolytic (plasminogen activator inhibitor-1 and α2-antiplasmin), and inflammatory (α1-antitrypsin [α1-proteinase inhibitor]) pathways. Serpins inhibit their target enzymes via a suicide-substrate inhibition mechanism that requires full function of the target proteinase catalytic apparatus and involves the formation of a covalent complex between the enzyme active site serine and the serpin.16 Serpins are globular molecules with protruding reactive center loops (RCLs) containing substrate-like sequences that serve as “bait” for their target enzymes (FIGURE 18.2A). Inhibitory complex formation begins when a target enzyme cleaves the scissile P1-P1′ bond (nomenclature of Schechter and Berger17) of the RCL. This generates an acyl-enzyme complex in which the P1 residue is covalently linked to the target proteinase active site serine, and in which the polypeptides generated by reactive loop cleavage become mobile and are rearranged.18,19 The P1-containing polypeptide incorporates as strand 4A in central β-sheet A of the serpin, translocating the covalently bound target enzyme approximately 70 Å to the opposite pole of the inhibitor.20 Structures of the canonical serpin α1-antitrypsin in inhibitory complexes with trypsin21 and elastase22 (FIGURE 18.2B) show that inhibitory complex formation results from critical protein conformational changes in both the inhibitor and the target enzyme. The P1-P1′ bond of the RCL is cleaved by the target enzyme, which promotes a large-scale protein conformational change, wherein the stressed, five-stranded A-sheet metastable conformation relaxes into a hyperstable, sixstranded A-sheet conformation in which the acylated enzyme is translocated to the opposite pole of the serpin. The translocated target enzyme molecule is crushed against the hyperstable serpin molecule in such a way that distortion of catalytic triad geometry prevents deacylation and regeneration of active proteinase.
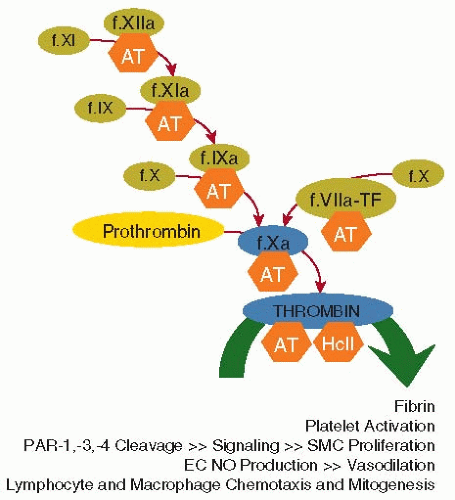 FIGURE 18.1 ATIII and HcII inhibition of coagulation pathway enzymes. ATIII inhibits coagulation factors in the intrinsic, extrinsic, and common pathways. ATIII and HcII inhibit thrombin, which has coagulant and noncoagulant functions. |
Heparin Cofactor Activation of Antithrombin III
ATIII exhibits the general serpin structural and mechanical features described above, but it is somewhat unusual in being a considerably less efficient inhibitor than typical members of this family. Whereas rate constants for target proteinase inhibition by most serpins are in the 106 to 107 M-1 sec-1 range, the values for ATIII inhibition of its target enzymes are approximately three orders of magnitude lower. In contrast to other serpins, which have fully exposed RCLs, native ATIII circulates in a conformation in which the amino-terminal end of its RCL is partially inserted into its central β-sheet A12,14 (see P14 residue S380 of orange RCLs of native and intermediate ATIII in panels a and b of FIGURE 18.3). RCL insertion was originally thought to reduce ATIII inhibition rates by limiting target enzyme access to the arginine393 P1-P1′ scissile bond (blue sphere on orange RCL).23 However, it is now appreciated that native conformation presentation of an exosite (cyan surface under orange RCL in FIGURE 18.3), which comprises elements that positively and negatively influence interactions with factors Xa and IXa, contributes substantially to reduced ATIII inhibition rates in the absence of cofactor heparin.24,25
Heparin and HSPGs containing a very specific pentasaccharide sequence serve as pharmaceutical and physiologic cofactors for ATIII and activate it to achieve inhibition rates comparable to those that most other serpins are capable of without cofactors. The specific pentasaccharide sequence26,27 that mediates high-affinity binding and activation of ATIII has the structure illustrated in FIGURE 18.4. This pentasaccharide is present in the approximately 30% of pharmaceutical heparin molecules having anticoagulant activity28 and in vascular wall and matrix HSPGs, which are endogenous ATIII receptors and activators.29,30
ATIII thrombin, FXa, and FIXa inhibition rates are accelerated up to several thousand-fold by a combination of heparin-dependent conformational change and bridging mechanisms. The relative importance of the conformational change and approximation mechanisms varies with the target enzyme.
A bridging effect is the most important factor in the activation of thrombin inhibition. Thrombin inhibition is accelerated just twofold by pentasaccharide, whereas pentasaccharide-containing heparin chains of >18 sugar units in length accelerate the rate of inhibitory complex formation by more than 1,000-fold, using an approximation mechanism wherein different regions of the same extended heparin chain bind to the pentasaccharide-binding site on ATIII and to anion-binding exosite II on thrombin.31,32
In contrast, an allosteric induced fit conformational change mechanism plays an important role in the acceleration of ATIII FXa and FIXa inhibition by heparin. Pentasaccharide alone accelerates rates of FXa and FIXa inhibitory complex formation by approximately 300-fold.28,33,34 In the presence of Ca2+, full-length heparin produces further inhibition rate increases, revealing that in addition to the primary conformational change-based mechanism of FXa and FIXa activation, there are also template contributions to full-length heparin cofactor-mediated inhibition of these targets.33,34,35 The FXa/FIXa exosite has been mapped to a region of β-sheet C (cyan in FIGURE 18.3) located under the RCL loop (orange in FIGURE 18.3). The protein conformation of the exosite as it is presented in native ATIII is unfavorable for interactions with the target enzymes. However, cofactor binding to the heparin-binding site of ATIII induces long-range allosteric conformational changes that expose exosite features that promote recognition and interaction with factors Xa and IXa and minimize repulsive interactions that were present in the native conformation exosite.24,25,36,37
As illustrated in FIGURE 18.3, the ATIII pentasaccharide-binding site is located at the junction of the N-terminal polypeptide (ruby) with helices A (light yellow) and D (pink), some 30 Å distant from the RCL (orange). The pentasaccharide is drawn in stick representation with blue carbon, red oxygen, yellow sulfur, and dark blue nitrogen atoms. Crystal structures of AT*H5 complexes8,9,12 allowed direct identification of ATIII surface residues that contact the cofactor and largely confirmed heparin-binding site residue assignments previously made on the basis of early chemical modification studies and the analysis of naturally occurring, thrombosis patient ATIII mutants with heparin-binding defects. Detailed information about the roles of specific residues and their quantitative contributions to the binding of the heparins and to the stabilization of the high-affinity AT*H complexes have more recently become available from investigations of recombinant mutant ATIIIs.
Heparin binding to and activation of ATIII has been described in terms of the two-step, three-state mechanism presented in FIGURE 18.5.38 An initial weak binding complex, AT-H, forms first. Then, in a second step, a protein conformational change converts the AT-H intermediate to the activated, high-affinity AT*H complex that is competent for rapid FXa and FIXa inhibition. The overall affinity (Kd) of antithrombin for heparin results from the combined effects of the K1 binding and k+2 and k-2 rate constants defined in the two-step mechanism. K1 represents the dissociation equilibrium constant for formation of the initial weak binding complex, AT-H. k+2 measures the rate for
conformational conversion of initial weak AT-H complex to the high-affinity AT*H complex. Finally, k-2 represents the reverse rate of the conformational change (i.e., the stability of the AT*H complex). The extent to which individual ATIII residues and pentasaccharide components contribute to each of the above steps was determined by measuring the binding equilibrium and kinetic constant values for ATIII mutants and truncated pentasaccharides and by comparing them to those of ATIII and pentasaccharide controls.
conformational conversion of initial weak AT-H complex to the high-affinity AT*H complex. Finally, k-2 represents the reverse rate of the conformational change (i.e., the stability of the AT*H complex). The extent to which individual ATIII residues and pentasaccharide components contribute to each of the above steps was determined by measuring the binding equilibrium and kinetic constant values for ATIII mutants and truncated pentasaccharides and by comparing them to those of ATIII and pentasaccharide controls.
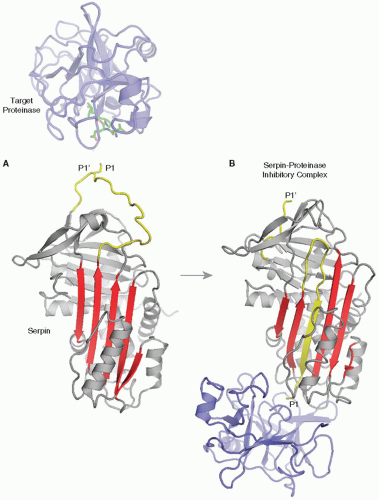 FIGURE 18.2 Serpin inhibition mechanism. A: The native serpin has a five-stranded central β-sheet A (red) and an exposed RCL (yellow). A substrate-like sequence in the RCL is recognized by a target enzyme (blue), which initiates cleavage of the P1-P1′ scissile bond. B: Formation of an inhibitory complex between the serpin and target proteinase involves (a) recognition and cleavage of the P1-P1′ peptide bond in the RCL polypeptide, (b) formation of a covalent acyl linkage between the serpin P1 residue and the enzyme active site serine, (c) expansion of the serpin central β-sheet by incorporation of the cleaved RCL polypeptide as a 6th strand, (d) translocation of the P1 residue and covalently bound enzyme from the north to the south pole of the serpin molecule, and (e) deformation of active site geometry in the translocated proteinase, which prevents deacylation and regeneration of active enzyme. Drawings were generated from native α1-proteinase inhibitor (1HP7.pdb), pancreatic elastase (1H9L.pdb), and covalent α1-PI-PPE covalent complex (2D26.pdb) structures using PyMol. |
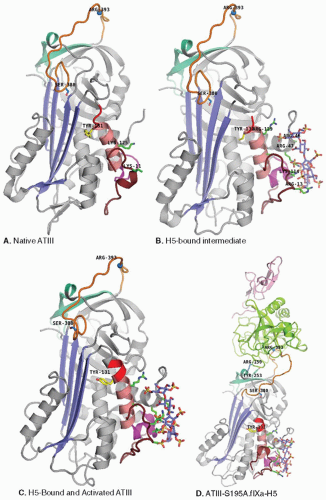 FIGURE 18.3 Heparin-mediated induced-fit allosteric activation of ATIII. (a) Native ATIII (1E04.pdb), (b) pentasaccharide-bound intermediate (1NQ9.pdb), (c) pentasaccharide-bound and activated ATIII (1E05.pdb), (d) ATIII-S195A.FIXa-pentasaccharide complex (3KCG.pdb). Drawings were generated from the indicated pdb files using PyMol. Color coding: β-sheet A is slate blue and the RCL is orange. P1 Arg-393 and P14 Ser-380 residues of the RCL are indicated by a marine blue sphere and a marine blue side chain, respectively. The β-sheet C region comprising the ATIII exosite for factors IXa and Xa is cyan. Structural elements forming the heparin-binding site are drawn in shades of reds and yellow: helix D pink; helix P magenta; N-terminal polypeptide ruby; C-terminal helix-D extension polypeptide red; helix A light yellow. Side chains of basic residues of the heparin-binding site are shown as green-element sticks. Pentasaccharide is drawn with blue-element sticks. Conformational changes induced by H5 binding to antithrombin can be visualized by comparing panels a, b, and c: Native ATIII (a) is a poor inhibitor due to suboptimal presentation of FXa/FIXa exosites (cyan) and the presence of repulsive sites in the surface surrounding the RCL (orange). Heparin allosterically activates ATIII by binding to site that is on the other side of the molecule. The cofactor initially binds to K11 and K125 of the native conformation (green side chains in panel a). Induced-fit conformational changes in both the H5 and ATIII molecules promote K114, R13, R46, R47, and R129 interactions with the cofactor and the formation of helix P (magenta) (see panel b). These binding interactions extend helix D at its C-terminal end (red), which causes tyrosine-131 rotation (compare positions of yellow Y131 side chain in panels a, b, and c). Exposure of buried Y131 ring leads to consolidation of the serpin core and long-distance changes in the electrostatics and H-bond network of the surface that underlies the RCL. The ATIII exosite for FXa and FIXa (cyan) is rearranged in a way that promotes favorable interactions and alleviates negative interactions with these target enzymes. See chapter text for greater detail. Structure of encounter complex of S195A-FIXa with H5-activated ATIII: FIXa heavy chain is green and light chain is pink. Pentasaccharide binding has induced activating protein conformational changes that elongate helix D and rotate tyrosine-131 to promote closure of central β-sheet A, P14 S380A expulsion, and favorable interactions of the exosite surface and key residue Y253 (cyan side chain) with R150 (blue side chain) of FIXa, and P1 arginine393 (blue sphere) with the S195A-FXa “catalytic” triad. |
K1 analysis showed that initial recognition and low-affinity binding of the pentasaccharide to native ATIII is mediated by its DEF nonreducing end (see FIGURE 18.4), which interacts with K125 from helix D and K11 in the N-terminal polypeptide.39,40,41,42,43 Side chains of the two basic residues that participate in the initial binding step are drawn and labeled in the native structure of FIGURE 18.3A.
The weak binding interaction of the first step leads to conformational adjustments in both the pentasaccharide and the antithrombin molecules. Pentasaccharide DEF complementarity with ATIII is enhanced and creates a complementary secondary site for binding a preferred conformer of the GH disaccharide.40,41 On the antithrombin side, the protein conformational change converting AT-H to AT*H requires K114, K125, and F122,39,44,45 which make the largest contributions to k+2 and mediate the formation of a new helix P (magenta in FIGURE 18.3) when pentasaccharide is bound. The activated AT*H complex that forms is stabilized by interactions requiring F122, R129, and, especially, K114, as evidenced by large increases in k-2 when these residues are mutated.39,44,45 Side chains of basic residues that participate in the second binding step leading to further increases in heparin affinity, and ultimately long-distance conformational changes that mediate exosite presentation, are drawn and labeled in the intermediate structure of FIGURE 18.3B.39,40,41,42,43
Helix-D elongation (see red polypeptide at the C-terminal [top] end of helix D [pink] in FIGURE 18.3) also contributes to increased affinity for heparin and allosteric activation of FXa inhibition. ATIII mutants in which helix-D elongation is inhibited by deletion(s)46 or by proline introduction47 in the elongated helix-D region exhibit decreased pentasaccharide- and heparin-binding affinities and cannot be fully activated for FXa inhibition. Conversely, replacement of helix-D residue Y131 with a leucine produces 30-fold activation of FXa inhibition in the absence of cofactor and increased affinity for heparins.48 Thus, the rotation of helix-D residue Y131 (compare positions of yellow Y131 side chain in panels a to d of FIGURE 18.3), caused by occupancy of the heparin-binding site or by Y131 phenol side chain substitution with smaller, nonphenyl groups, contributes to allosteric activation of ATIII by promoting consolidation of the hydrophobic core underlying central β-sheet A and long-range protein conformational changes that are associated with rearrangement of bonding networks formed by residues in the A-sheet, the hF arm, and the reactive loop polypeptide,9,49 and more favorable presentation of the exosites for factors Xa and IXa recognition. FIGURE 18.3D shows the crystal structure for the encounter complex of S195A FIXa (green heavy chain, pink light chain) with H5-activated ATIII. Note that pentasaccharide binding has induced activating protein conformational changes that elongate helix-D and rotate tyrosine-131 to promote closure of central β-sheet A, P14 S380A expulsion, and favorable interactions of the exosite surface and key residue Y25336,50 with arginine-150 of FIXa, and P1 arginine393 with the S195A-FXa “catalytic” triad.10
The K1 binding and k+2 and k-2 rate constants from the two-step mechanism of FIGURE 18.5 together determine the overall affinity of the ATIII-pentasaccharide interaction. At physiologic pH and at ionic strength, the Kd for binding of the major α-ATIII isoform of human plasma to pentasaccharide and longer chain high-affinity (i.e., pentasaccharide-containing) heparin molecules are approximately 50 and 20 nM, respectively, as determined by both binding equilibrium and binding kinetic measurements.51 Measurements of the free energy of binding with pentasaccharide for mutants of key heparin-binding site residues have determined that lysine114 and phenylalanine122 make the greatest contributions to the binding energy.39,45 Lysine125 and arginine129 also make significant contributions,42,44 whereas lysine11 and arginines13 and 47 make only small contributions.43,52 Analysis of binding energy losses across the mutant series also indicates that cooperative interactions between combinations of the previously described residues contribute to high-affinity cofactor binding.53
The binding of pentasaccharide and ATIII is mediated by ionic as well as by nonionic interactions. From the salt dependence of the Kd, it has been determined that ionic interactions of the pentasaccharide and the six basic residues on ATIII are responsible for approximately 40% of the free energy of binding.28,32,39,42,43,44,52 Nonionic interactions (i.e., hydrogen bonds and van der Waals and hydrophobic interactions) contribute approximately 60% of the binding free energy, with centrally
located phenylalanine122 being responsible for a substantial share by positioning R47 and K114 for extensive contact with the cofactor and bridging structural elements involved in forming and stabilizing the high-affinity conformation.45
located phenylalanine122 being responsible for a substantial share by positioning R47 and K114 for extensive contact with the cofactor and bridging structural elements involved in forming and stabilizing the high-affinity conformation.45
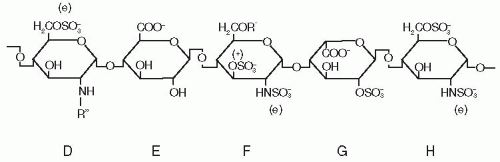 FIGURE 18.4 Structure of the heparin pentasaccharide that binds ATIII with high affinity and activates it for efficient target proteinase inhibition. Structural variants are indicated by R′ (-H or -SO3–) and R″ (-SO3– and -COCH3). The 3-O-sulfate group marked by an asterisk is unique to the antithrombin-binding region of the heparin molecule. In addition, the three sulfate groups indicated by (e) are essential to the high-affinity binding of antithrombin. (Modified from Tollefsen, DM, Arterioscler Thromb Vasc Biol 2007;27:456, with permission.) |
Endogenous Activation of Antithrombin III
As previously noted, native conformation ATIII molecules circulating in the blood have limited inhibitory activity. They become activated, however, upon binding to vessel wall HSPGact molecules (3-O-sulfated pentasaccharide-bearing anticoagulantly active HSPGs), which serve as the physiologic cofactor of ATIII. HSPGact is present on vascular endothelium and in the underlying matrix and serves to localize and concentrate activated antithrombin on and in the vessel wall. It is generally believed that activated ATIII molecules bound to vascular surface HSPGact receptors contribute substantively to the anticoagulant and antithrombotic properties of the endothelium by inhibiting free thrombin, FXa, and other activated coagulation-factor molecules that are generated by membrane-bound activation complexes and released into the diffusion layer at the blood vessel-wall interface.
Physiologic activation of ATIII by a vascular component was initially investigated in a rat hindlimb quarters model perfused with ATIII and thrombin. A tightly bound, heparinase-sensitive component accelerated the rate of thrombin-antithrombin complex formation 15- to 19-fold.30 Further studies with normal versus mast cell-deficient mice established that the accelerating vascular component was HSPGact rather than a mast cell-derived heparin species.54 This anticoagulantly active HSPGact species constitutes about 5% of the total HSPG associated with rat microvascular endothelial cells.55 The integral membrane proteins ryudocan and syndecan serve as core proteins for both HSPGact and HSPGinact (anticoagulantly inactive HSPG).56 Ryudocan and syndecan have, respectively, three and five extracellular serine attachment sites that are posttranslationally modified with glycosaminoglycans (GAGs) composed of alternating N-acetylglucosamine and glucuronic acid/iduronic acid residues. The copolymer is further modified by multiple sulfation reactions to generate heparan sulfate chains with regions of varying structure. The presence of chains bearing pentasaccharide sequence(s) with essential 3-O-sulfation (See FIGURE 18.4) confers HSPGact status on a small subset of HSPG molecules.
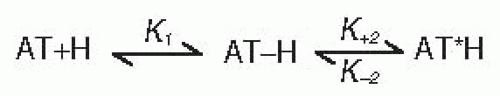 FIGURE 18.5 Two-step mechanism for heparin binding to and activation of ATIII. AT, antithrombin III; H, heparin; AT-H, initial weak antithrombin-heparin intermediate; K1, dissociation equilibrium constant for AT-H formation; AT*H, high-affinity antithrombin-heparin complex which is competent for rapid FXa inhibition; k+2, rate of conformational conversion of the weak AT-H intermediate to the high-affinity AT*H complex; k-2, reverse rate of the conformational change. |
The anatomic localization of HSPGact has been studied using immunohistochemistry and radiolabeled ATIII perfusion techniques. ATIII localization was investigated using immunohistochemistry techniques in biopsies of normal human hearts before transplantation.57 Venous endothelium, but not capillary endothelium, stained positively for ATIII. ATIII was also present around smooth muscle cells and around intima of small arteries and arterioles, but not around large arteries, in apparent contradiction to the results of subsequent studies in which normal rat aorta was perfused with 125I-labeled ATIII. These perfusion studies29 demonstrated the presence of ATIII-binding HSPGact on and under the aortic endothelium. Small amounts of HSPGact (1%) were observed on the luminal side of aortic endothelial cells, whereas the subendothelial basement membrane and extracellular matrix on the abluminal side of the endothelium contained much greater amounts of ATIII-binding HSPGact. The observed distribution suggests that luminal HSPGact may provide a basal level of activated ATIII for scavenger inhibition of activated coagulation enzymes exposed to the endothelium, and that these basal levels may be dramatically increased in injured regions following endothelial damage and liberation of the larger pool of abluminal HSPGact and its associated bound and activated ATIII.
The physiologic importance of ATIII binding to endogenous vascular surface HSPGact is supported by the occurrence of lethal thrombosis in mice homozygous for an ATIII mutation that reduces binding to heparin/HSPG.58 Similarly, thrombosis was prevented in stents coated with the high-affinity, pentasaccharide-containing fraction of heparin (which binds antithrombin) but not in stents coated with low-affinity,
pentasaccharide-depleted heparin (which does not support ATIII binding).59 Therefore, antithrombin-HSPG interactions play a crucial role in maintaining circulatory system patency. A previous contrary report based on the nonthrombotic phenotype of mice deficient for 3-OST-160 may reflect redundancy of enzymes that mediate 3-O-sulfation of the pentasaccharide sequence of HSPG.61,62
pentasaccharide-depleted heparin (which does not support ATIII binding).59 Therefore, antithrombin-HSPG interactions play a crucial role in maintaining circulatory system patency. A previous contrary report based on the nonthrombotic phenotype of mice deficient for 3-OST-160 may reflect redundancy of enzymes that mediate 3-O-sulfation of the pentasaccharide sequence of HSPG.61,62
Finally, in addition to vascular HSPGs, the endothelial cell thrombin receptor thrombomodulin can bind ATIII and enhance its inhibition of receptor-bound thrombin by a factor of approximately 8.63 This thrombin-specific effect is mediated by a highly sulfated chondroitin sulfate chain on thrombomodulin,64,65 which interacts with both thrombin (at exosite II,66) and with ATIII. Chondroitin sulfate occupancy of exosite II of anticoagulant thrombin (i.e., thrombomodulin-bound thrombin) blocks heparin binding to the enzyme and precludes heparin/ATIII inhibition at the very rapid rates observed for procoagulant thrombin.67
Pharmacologic Activation of Antithrombin III
The clinical use of heparin as an anticoagulant began more than 60 years ago.68 The name heparin derives from the fact that the anticoagulant was originally extracted from liver; however, commercial preparations are now usually obtained from pig intestinal mucosa or bovine lung, which are good sources of mast cells. Mast cell heparin proteoglycan is composed of a serglycin core protein modified with 60 to 100,000 Da heparin sulfate GAG chains.69 The heparin proteoglycan polysaccharide chains are intracellularly cleaved to form smaller fragments (7,000 to 25,000 Da) that are present in pharmaceutical heparin. Standard pharmaceutical heparin is a heterogeneous mixture of polysaccharide molecules (average 15,000 to 18,000 Da). In addition to a high degree of size/length heterogeneity, there is also a significant amount of compositional heterogeneity. Typically, only one-third of the molecules in a standard pharmaceutical heparin preparation bear the ATIII-binding pentasaccharide sequence and exhibit anticoagulant activity.
Unfractionated heparin is used widely in clinical settings. It is given to prevent and treat venous thrombosis and for the management of arterial disease. Intravenous administration of heparin doses that prolong the activated partial thromboplastin time (aPTT) to 1.5 to 2.5 times control values is highly effective for the prevention and treatment of venous thrombosis.70 Heparin is also widely utilized in extracorporeal circulation, for flushing indwelling catheters, for coating biomaterials and medical devices, and in many other hospital procedures. However, there are major drawbacks associated with the use of unfractionated heparin, including increased risk of hemorrhage and thrombocytopenia, variable patient response, and prolonged hospitalization because of the need for anticoagulant monitoring and dosing adjustments. In addition to inhibiting clotting enzyme function, heparin also interferes with platelet function, which contributes to its hemorrhagic side effects. On the basis of studies with size-fractionated heparins, which indicated that the high molecular weight fraction (average 20,000 Da) was considerably more active at inducing platelet aggregation than was the low molecular weight fraction (average 7,000 Da),71 low molecular weight heparins (LMWHs) were suggested as a potentially advantageous alternative to standard heparin.72
Several LMWHs have been developed and evaluated in clinical trials and are now frequently used in hospital and outpatient settings. LMWHs are derived from unfractionated heparin by enzymatic or chemical cleavage. Their range of fragment size is from 1,800 to 12,000 Da, with an average of around 5,000 Da. As a result of their small fragment size and the cleavage into smaller pieces of most chains that were long enough (>18 sugars) to approximate (bridge) ATIII and thrombin, LMWHs preferentially inhibit factor Xa and have antifactor Xa to antithrombin ratios of 2:1 to 4:1 (vs. 1:1 for standard heparin). Compared with standard heparin, LMWHs have decreased affinity for endothelial cells73,74 and exhibit reduced nonspecific binding to heparin-binding proteins.75 These properties improve bioavailability and produce a more predictable dose response. The improved pharmacokinetic profile permits LMWHs to be administered subcutaneously once or twice daily on a weight-adjusted basis without laboratory monitoring. These attributes are an important advantage over standard heparin, which has a highly variable patient response that necessitates frequent laboratory monitoring, dose adjustment, and prolonged hospitalization. Clinical trials have established that for eligible patients with deep vein thrombosis, outpatient, unmonitored, subcutaneous, weight-adjusted LMWH is as effective and as safe as inpatient treatment with intravenous, unfractionated heparin and affords considerable cost reductions.76
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


