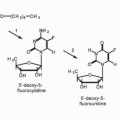
The vinca alkaloids are naturally occurring or semisynthetic nitrogenous bases that are present in minute quantities in the pink periwinkle plant
Catharanthus roseus G. Don (formerly
Vinca rosea Linn). The early medicinal uses of
C. roseus for controlling hemorrhage, scurvy, toothache, and diabetes, and for the healing of chronic wounds led to the screening of these compounds for their hypoglycemic activity, which turned out to be of little importance compared with their anticancer properties.
41,
42,
43,
44,
45,
46,
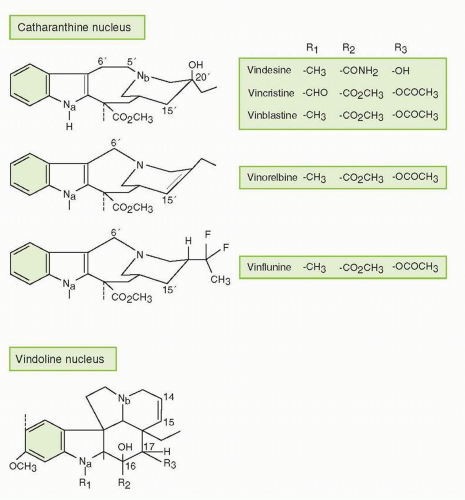
47 Although many vinca alkaloids have been investigated clinically, only VCR, VBL, and vinorelbine (VRL) are approved for use in the United States. A third widely studied vinca alkaloid, vindesine (VDS, desacetyl VBL carboxyamide), a semisynthetic derivative and human metabolite of VBL, was introduced in the 1970s. It has been used in combination with other agents, particularly the platinating agents and/or mitomycin C (or both), to treat non-small cell lung cancer, but VDS is also active in other hematologic and solid malignancies.
7,
29,
43,
44 Although VDS demonstrated notable activity against several tumor types, particularly non-small cell lung cancer, it has been available only for investigational purposes in the United States and has not demonstrated a unique role in cancer therapeutics.
43,
44 The semisynthetic VBL derivative vinorelbine, VRL (5′-norhydro-VBL), which is structurally modified on its catharanthine nucleus, is approved in the United States as either a single agent or in combination with cisplatin to treat non-small cell lung cancer and has also been registered for advanced breast cancer in many other countries.
7,
29,
45,
46,
47,
48 In addition to demonstrating broad antitumor activity as a single agent and some evidence that it may not be completely cross-resistant with VCR and VBL, VRL can be administered orally, in contrast to other available vinca alkaloids.
49 More recently, vinflunine (VFL), which is a bifluorinated vinca alkaloid, has demonstrated notable clinical activity in bladder cancer, as well as preliminary activity in breast, lung, and other cancers, but despite these activities, unique roles for VDS and VFL relative to other vinca alkaloids have not yet been demonstrated.
50,
51,
52,
53,
54 The key features of these vinca alkaloids are listed in
Table 13-1.
Despite the minor structural differences between VCR and VBL, their antitumor and toxicologic profiles differ greatly. VCR is used more commonly in pediatric oncology than in adults with cancer, most likely owing to the higher level of sensitivity of pediatric malignancies and better tolerance of therapeutic VCR doses in children. VCR is an essential part of the combination chemotherapeutic regimens used for acute lymphocytic leukemia and plays an important role in the treatment of non-Hodgkin’s lymphoma. VCR-based combination regimens, particularly those in which VCR is administered with doxorubicin and dexamethasone (known as VAD) and occasionally other newer agents such as bortezomib, are used to treat multiple myeloma.
29 VCR also plays a role in the treatment of Wilms’ tumor, neuroblastoma, medulloblastoma, osteosarcoma, Ewing’s sarcoma, and other types of pediatric sarcoma, and small cell lung cancer.
29,
41,
42,
44 The agent has also been used as a component of a combination regimen consisting of procarbazine, lomustine, and VCR (known as PCV) in the neoadjuvant, adjuvant, and advanced settings of several types of uncommon brain malignancies anaplastic including oligoastrocytoma and oligodendroglioma.
55 In addition, VCR and VCR-loaded platelet transfusions are used occasionally to treatment refractory autoimmune thrombocytopenia, and VCR has been successfully used to treat other nonmalignant immunologically mediated disorders such as autoimmune hemolytic anemia, hemolytic uremic syndrome, thrombotic thrombocytopenia purpura, and steroid-dependent nephrotic syndrome.
29,
41,
42,
56 VBL has been an integral component of combination therapeutic regimens for germ-cell malignancies (with cisplatin and bleomycin, PVB) and Hodgkin’s disease (with doxorubicin [adriamycin], bleomycin, and DTIC, ABVD) and has been used in combination with other agents to treat Kaposi’s sarcoma and bladder, brain, and non-small cell lung and breast cancers.
29 In addition to the clinically relevant antitumor activity of VRL in non-small cell and breast cancers, VRL has demonstrated activity in advanced ovarian carcinoma and lymphoma, but a unique role in the treatment of these cancers has not been defined.
29,
47 It has also been reported that VRL as single-agent treatment confers reasonable therapeutic benefit in elderly patients with advanced breast and lung cancers.
29,
46,
47,
48
Mechanism of Action
The vinca alkaloids induce cytotoxicity by interacting with tubulin and disrupting microtubule function.
2,
3,
5,
6,
7,
29,
30,
41,
42,
47,
50,
51,
52,
53,
57,
58,
59 However, they affect other biochemical and biologic actions that may or may not be related to their effects on microtubules, such as the intracellular transport of amino acids; syntheses of RNA, DNA, and proteins; lipid metabolism; glutathione oxidation; cellular glycolysis; cellular release of hormones and pharmacological substances (e.g., antidiuretic hormone, histamine, epinephrine); calcium-calmodulin-regulated cAMP; and the integrity of the cellular membranes.
7,
27,
28,
29,
41,
47 Many of these effects occur only after treatment with superpharmacological concentrations that are not readily attained in vivo, whereas nanomolar concentrations, which are readily achieved in clinical practice, induce typical antimicrotubule effects. Although the vinca alkaloids preferentially disrupt proliferating cells and tissues, they also affect nonproliferating tissues that are rich in tubulin such as neurons and platelets.
60The cytotoxic actions of the vinca alkaloids are principally due to their effects on the mitotic spindle apparatus. In support of this mechanism, there is a strong relationship between cytotoxicity and the dissolution of the mitotic spindle.
46 Furthermore, the accumulation of mitotic figures correlates with both drug concentration and duration of treatment. However, the vinca alkaloids and other antimicrotubule agents also perturb both malignant and nonmalignant cells in the nonmitotic cell-cycle phases, which is not surprising since microtubules are involved in many nonmitotic functions, including chemotaxis, migration, intracellular transport, movement of organelles, secretory processes, membrane trafficking, and transmission of growth factor signals from the cell surface receptor to the nucleus.
15,
61,
62The vinca alkaloids bind rapidly, avidly, and reversibly to sites on tubulin, known as the vinca domain, which appears to be the same binding sites for plant alkaloids such as maytansine, but distinct from those of the taxanes, GTP/GDP, and the site on the tubulin heterodimer shared with colchicine, podophyllotoxin, steganacin, combretastatin, and many synthetic compounds.
2,
3,
5,
7,
30,
31,
41,
51,
53,
54,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70 Unlike colchicine, the vinca alkaloids bind directly to microtubules without first forming a complex with soluble tubulin, and the vincas do not copolymerize with the tubulin lattice of the microtubule.
2,
3,
5,
65,
66,
67,
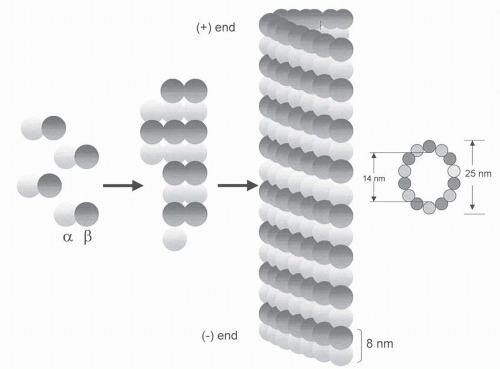
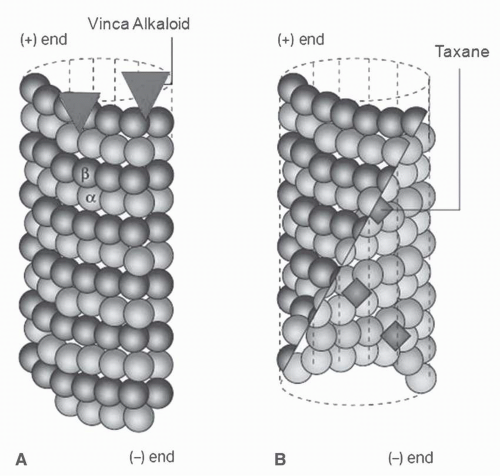
68The vinca alkaloids bind to microtubules at two binding sites, each with different affinities including high affinity sites (
Kd, 1 to 2 μmol) located at the ends of microtubules and low affinity sites (
Kd, 0.25 to 0.3 mmol) sites located along the sides of microtubule surfaces (
Fig. 13-3).
3,
6,
65,
66,
67,
68 The binding of the vinca alkaloids to high-affinity sites is responsible for the substoichiometric and potent suppression of tubulin exchange that occurs at low vinca alkaloid concentrations (<1 μmol). This engagement disrupts treadmilling, dynamic instability, and other dynamic processes, but microtubule mass is not affected. Low concentrations of the vinca alkaloids strongly enhance dynamic instability at the minus end of microtubules, whereas dynamic instability is inhibited at the plus end. These actions increase the time that microtubules spend in a state of attenuated activity, neither growing nor shortening, the end result of which is a potent block at the metaphase/anaphase boundary in mitosis.
2,
3,
5,
55,
56,
65,
66,
67,
68The binding of the vinca alkaloids at high stoichiometric concentrations (μmol) to low-affinity sites (
Kd, 0.25 to 3.0 mmol) along the sides of microtubules is accompanied by reduced microtubule mass due to tubulin depolymerization. This type of binding induces tubulin to self-associate into nonmicrotubule tubulin polymers and ordered aggregates through a self-propagation pathway. Self-propagation occurs as vinca alkaloid binding progressively weakens the lateral interactions between protofilaments, induces conformation changes in tubulin, and exposes new sites. The exposure of new sites further increases the binding affinity of the vinca alkaloids and, in turn, results in the formation of vinca alkaloid-tubulin spiral aggregates, protofilaments, and paracrystalline structures, which ultimately results in the disintegration of the microtubules. MAPs stabilize the longitudinal interactions between dimers in the proto-filaments as they splay apart after binding to the vinca alkaloid, as illustrated in
Figure 13-4.
69 MAPS may also mediate the effects of the vinca alkaloids. For example, VBL increases the affinity of microtubules for the MAP stathmin, which destabilizes microtubules.
71The vinca alkaloids induce a block in mitosis at the metaphase/anaphase transition.
2,
3,
6,
7,
27,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70 Following nuclear envelop breakdown, the vinca alkaloids block mitotic spindle formation and reduce tension at the kinetochores of the chromosomes. Although chromosomes may condense, they remain scattered in the cells. The chromosomes separate along their lengths but still remain attached at their centromeres.
66,
70 Following vinca alkaloid treatment, mitotic progress is delayed in a metaphase-like state with chromosomes “stuck” at the spindle poles, unable to move to the spindle equator.
The cell-cycle signal to the anaphase-promoting complex, which is required for the cell to transition from metaphase to anaphase, is blocked and the cells eventually undergo apoptosis. However, cyclin B concentrations may remain high and cell-cycle progression to interphase in the absence of anaphase or cytokinesis may occur, resulting in chromatin decondensation and formation of multilobed nuclei.
2,
3,
6,
64 Although treatment of cells with low concentrations of the vinca alkaloids may result in the disruption of spindle microtubule dynamics without microtubule depolymerization in mitotic cells, it can nonetheless lead to apoptosis, which involves the inactivation of antiapoptotic factors and induction of proapoptotic factors (see the sections on “
Mechanism of Action” and “
Mechanisms of Resistance” under “
Taxanes”).
2,
3,
6,
7,
22,
26,
51,
52,
54,
70,
72,
73,
74,
75,
76,
77,
78The processes that govern whether a cell dies during mitosis or exits mitosis in the presence of an antimitotic drug are not entirely clear. Several fates have been described for cells that exit mitosis in the presence of an antimitotic drug, including cell-cycle arrest, apoptosis, and cell-cycle progression. Although the molecular factors that govern these fates are not well understood, there is abundant evidence that p53 is involved, perhaps by restraining cell-cycle progression following exit from a prolonged mitotic block.
6,
26,
73,
74,
75,
76,
77 It is also becoming increasingly clear that the fate of the cell in response to drug is determined not only by events occurring during mitotic arrest but also by the consequences of events after mitotic exit, perhaps involving signaling pathways and cellular machinery that are principally operative during interphase including cdk1, cyclin B, p21 and various apoptosis proteins.
6,
26,
73,
74,
75The relationships between the antiproliferative actions of the vinca alkaloids and various relevant subcellular effects, such as mitotic arrest, mitotic spindle disruption, and microtubule depolymerization, have been characterized in a series of elegant studies.
59 The antiproliferative effects of the vincas strongly related to the induction of both mitotic spindle disruption and metaphase arrest. These effects occur at the lowest effective drug concentrations with little or no microtubule depolymerization or disorganization of the mitotic spindle apparatus. With increasing drug concentrations, the organization of microtubules and chromosomes in arrested mitotic spindles deteriorates in a manner that is common to all vinca derivatives. The cumulative body of evidence data indicates that the antiproliferative effects of the vinca alkaloids at their lowest effective concentrations are caused by alterations in the dynamics of tubulin addition and loss at the ends of mitotic spindle microtubules rather than by depolymerization of the microtubules. Similar effects have been demonstrated with nocodazole, podophyllotoxin, and the taxanes.
72,
76,
78,
79In addition to their direct cytotoxic effects on tumor cells, the vinca alkaloids and other antimicrotubule agents disrupt malignant angiogenesis with surprising potency.
75,
80,
81,
82,
83 In vitro, 0.1 to 1.0 pmol/L VBL blocks endothelial proliferation, chemotaxis, and spreading on fibronectin, all essential steps in angiogenesis.
80,
82 However, the relative contribution of these antiangiogenic effects to the antitumor activity of the vinca alkaloids in the clinic is unclear.
Additionally, the vinca alkaloids possess radiosensitizing properties in vitro related to their ability to induce cell-cycle block in the G
2/M phase.
84,
85
Mechanistic and Functional Differences
With regard to the disruptive effects of the vinca alkaloids on microtubule dynamics, the naturally occurring vinca alkaloids VCR and VBL, the semisynthetic analog VRL, and the bifluorinated analog VFL impart similar actions, but they have distinguishing features as well.
54,
75,
86,
87 The vinca alkaloids differ in their tubulin-binding affinities: VCR > VBL > VRL > VFL.
51,
54 Consequently, the interaction between VFL and tubulin appears to be the most reversible, as shown by a greater reversibility of centrosome segregation after drug “wash-out” compared with the other vinca alkaloids, and this difference may be responsible, in part, for the differential effects of VFL on microtubule dynamics.
51,
54 However, the affinity for tubulin does not relate to the antitumor activity of the vinca alkaloid class. In fact, the relative tubulin-binding affinity may even be inversely correlated with their antitumor potencies in vivo.
51 Although VFL has a lower overall tubulin-binding affinity and a lower potential to induce vinca alkaloid-tubulin spiral polymers than VCR, VFL is more active than VCR in a variety of murine and human tumor models.
51,
54,
88,
89 Moreover, the effects of VFL and VRL on microtubule dynamics differ from those induced by VBL and VCR in that they not only decrease the microtubule growth rate; increase the mean duration of a growth event; and increase the percentage of time that microtubules spend time growing; but VFL and VRL decrease the time spent in attenuation to a much greater extent. In contrast, VBL and VCR decrease the shortening rate and increase the time microtubules spend in an attenuated state to a much greater extent than VRL and VFL.
51,
86 Suppression of microtubule treadmilling also occurs with VFL treatment, but to a lesser degree compared with VRL and VBL.
51,
86The explanation for the differential effects of the various vinca alkaloids on normal tissues and tumors is not clear. VCR, the most potent of the analogs in humans and the most neurotoxic, has the greatest affinity for tubulin.
51,
89 In contrast, VFL’s lower affinity for tubulin binding, as well as its greater potential intracellular sequestration, may contribute to its reduced incidence of peripheral neuropathy.
51 Peripheral neurotoxicity, possibly due to drug-induced microtubule loss, steroid hindrance of MAPs, and/or altered microtubule dynamics in axonal processes, is a common adverse effect of first-generation vinca alkaloids.
90 Although the vinca alkaloids may demonstrate similar potencies against preparations of tubulin derived from any given tissue, the differential sensitivities of various tissues to the vinca alkaloids are likely due to several factors.
59,
69,
87,
90,
91,
92,
93,
94,
95,
96 One possible factor is tubulin isotype composition, which is highly variable amongst tissues. Intracellular drug accumulation and tubulin binding vary according to tubulin isotype composition.
18,
19,
97 Neurons are enriched in
α-β-tubulin classes II and III, and the relatively high drug binding affinities for these isotypes may explain, in part, why the vinca alkaloids produce neurotoxicity.
88,
89,
97 The variable potencies of the vinca alkaloids with regard to the induction of tubulin spirals also appear to relate to their relative neurotoxic potencies.
88,
89,
97 In addition, the differences in the type and concentration of MAPs and posttranslational tubulin modifications between various tissues, which may influence drug interactions with tubulin, as well as differences in the cellular permeability and retention of the various vinca alkaloids, may affect the formation and stability of complexes formed between the vinca alkaloids and tubulin.
27,
59,
86,
89,
91,
92,
98,
99,
100 For example, the higher cellular retention of VCR compared with VBL in cultured leukemia cells may explain why VCR is more potent than VBL after brief treatment periods, whereas these effects of the vinca alkaloids differ to a lesser degree with more prolonged exposure times.
91,
96,
100,
101,
102,
103 Additionally, the vinca alkaloids directly inhibit palmitoylation of tubulin, and tubulin palmitoylation may relate to drug sensitivity.
104 The intracellular concentration of GTP concentrations may also influence the type of interactions between the vinca alkaloids and tubulin, and variable vinca alkaloid retention among tumors may relate to GTP hydrolysis.
101,
102,
103 Other factors that may explain why various tissues are differentially sensitive to the vinca alkaloid include differences in cellular pharmacology and pharmacokinetics, which is discussed in the next section.
Cellular Pharmacology
Although the vinca alkaloids are rapidly taken up into cells and accumulate intracellularly, steady-state intracellular/extracellular concentration ratios range from 5- to 500-fold depending on the cell type.
91,
93,
99,
103,
105 In murine leukemia cells, the intracellular concentrations of VCR are 5- to 20-fold higher than the extracellular concentrations, and this ratio has been reported to range from 150- to 500-fold for other vinca alkaloids in both human and murine leukemia cell lines.
96,
105,
106 In isolated human hepatocytes, VRL is more rapidly taken up and metabolized than other vinca alkaloids.
96,
105,
106,
107,
108 Although the vinca alkaloids are retained in cells for long periods and thus may have protracted cellular effects, there are marked differences in cellular retention amongst agents in this class.
109,
110,
111,
112,
113,
114 Overall, the most important determinant of drug accumulation and retention is lipophilicity, although a number of other factors undoubtedly play a role.
105,
106 Drug uptake and retention also appear to be determined by tissue-specific and drug-specific factors, as illustrated by studies indicating that the accumulation and retention of VRL in neurons are less than other vinca alkaloids (see the section on “
Mechanistic and Functional Differences” under “
Vinca Alkaloids”).
57,
102 Recent studies using cellulose-purified tubulin from porcine brain have demonstrated the following pattern of binding affinities: VCR > VBL > VRL > VFL, which mirrors the relative potential of the vinca alkaloids to induce tubulin spirals.
51,
98 As discussed in the last section, the differential tissue uptake of the vinca alkaloid appears to be related to the tissue composition of tubulin isotypes, each possessing different binding characteristics, uptake kinetics, efflux pumps, and intracellular reservoirs for drug accumulation. Although the binding affinity for tubulin appears to be less for VFL than for the other vinca alkaloids, its intracellular accumulation has been demonstrated to be higher than for VBL, VCR, and VRL, which may be due to the sequestration of VFL in, as of yet, undefined intracellular compartments and its slower release over time.
51,
75,
98Temperature-independent, nonsaturable mechanisms, analogous to simple diffusion, seem to account for most transport, and temperature-dependent saturable processes are less important.
27,
30,
74,
93,
105,
106 Although both drug concentration and treatment duration are important determinants of drug accumulation and cytotoxicity, the duration of exposure above a critical threshold concentration is perhaps the most important determinant of vinca alkaloid cytotoxicity.
96,
107 Cytotoxicity is directly related to the extracellular concentration of drug when the duration of treatment is kept constant; for prolonged exposure to VCR, the concentration yielding 50% inhibition ranges from 1 to 5 nmol/L.
107
Mechanisms of Resistance
Resistance to the vinca alkaloids develops rapidly in vitro with continuous exposure to these agents. Two types of mechanisms of resistance to the vinca alkaloids have been well characterized. The first mechanism is pleiotropic or multidrug resistance (MDR), which can be either innate (primary) or acquired. Although a large number of proteins mediate MDR, the best-characterized ones are the ATP-binding cassette (ABC) transporters, which transport a variety of substrates across cellular compartments and are encoded by a large transporter gene family.
108 These intracellular and extracellular membrane-spanning proteins transport endobiotics and xenobiotics across membranes and confer resistance to the vinca alkaloids, taxanes, and other structurally bulky, natural product chemotherapeutic agents in vitro. The most extensively studied ABC transporters with respect to conferring resistance to the vinca alkaloids are the permeability glycoprotein (Pgp), or the
MDR1 encoded gene product MDR1 (ABC Subfamily B1; ABCB1), and the multidrug resistance protein (MRP) (ABC Subfamily C2; ABCB1).
108,
109,
110,
111,
112,
113,
114,
115,
116,
117,
118MDR1 is a 170-kDa Pgp energy-dependent transmembrane transport pump that regulates the efflux of a large range of amphipathic hydrophobic substances, resulting in decreased drug accumulation. Pgp forms a channel in the membrane through which drugs are transported, and drug resistance is proportional to the amount of Pgp. Pgp is constitutively overexpressed by various normal tissues, including renal tubular epithelium, colonic mucosa, adrenal medulla, and other epithelial tissues. The efflux protein is also commonly
overexpressed by several human cancers, particularly those derived from tissues in which it is constitutively expressed (e.g., kidney and colon cancers). In the clinical setting, Pgp overexpression has been documented following treatment of patients with a variety of malignancies including lymphoma, leukemia, and multiple myeloma.
MDR1 confers varying degrees of cross-resistance to other structurally bulky natural products, such as the taxanes, anthracyclines, epipodophyllotoxins, actinomycin D (dactinomycin), and colchicine.
119,
120,
121,
122,
123,
124 These cells may have homogeneously stained chromosomal regions or double-minute chromosomes, which indicates the presence of an amplified gene that codes for Pgp.
110,
111 The specific Pgp associated with resistance to the vinca alkaloids shows slight antigenic amino acid sequence differences and a different peptide map after digestion than does Pgp from cells selected for resistance to colchicine or paclitaxel.
116,
122 In fact, two forms of the protein are produced by a single clone of VCR-resistant cells, and these forms undergo posttranslational modifications, particularly N-glycosylation and phosphorylation, which results in further structural diversity. This diversity may explain the greater degree of resistance for the specific agent used to induce resistance compared with other MDR substrates, and it also may explain the variable patterns of resistance among cells with the MDR phenotype. The composition of membrane gangliosides in cancer cells resistant to the vinca alkaloids has also been shown to differ from that of wild-type cells.
111 Although VFL is a substrate for Pgp, Pgp overexpression appears to be less involved in conferring resistance to VFL compared with other vinca alkaloids in various types of Pgpoverexpressing human cancers.
118,
125 The clinical ramifications of this resistance mechanism are not known, but VCR resistance, as assessed ex vivo, correlates with Pgp overexpression, particularly in childhood acute lymphoblastic leukemia (ALL).
112Resistance to the vinca alkaloids is also conferred by MRP1, which is a 190-kDa membrane-spanning protein that shares 15% amino acid homology with MDR1.
116,
117,
118,
119,
120,
121,
122,
123,
124,
125,
126,
127,
128 The expression of MRP1, a member of the ABC protein family distantly related to Pgp, is found in many types of cancer and has been implicated as being responsible for the MDR phenotype in cancers of the lung, colon, breast, bladder, and prostate, as well as leukemia.
116,
117,
118,
119,
120,
121,
122,
123,
124,
125,
126,
127,
128 Transient increase in MRP1 expression also correlates with resistance to MDR substrate drugs in cell lines transfected with
MRP1.123,
126 Amplification of
MRP1 has been identified in several laboratoryderived cancer cell lines with elevated levels of MRP1 protein, as well as increased energy-dependent drug efflux.
116,
117,
118,
128 MRP1 has been shown to transport glutathione conjugates of several types of compounds, including alkylating agents, as well as etoposide and doxorubicin, but it only confers resistance to the latter agents. The MRP1 profile also encompasses resistance to methotrexate but confers a low level of resistance, if any, to the taxanes and colchicine.
112,
113,
116,
118,
126,
127,
128 Also, MRP1 and other ABC transporters do not confer a significant resistance to VFL.
118,
125,
129 Expression of MRP in transfected or selected cell lines is principally localized to the plasma membrane and endoplasmic reticulum, suggesting that MRP1 mediates resistance by affecting drug sequestration and/or vesicular transport.
118,
127 Several other ABC transporters have also been characterized in vitro, including several that enhance cellular resistance to the vinca alkaloids; however, their roles in conferring inherent or acquired resistance to the vinca alkaloids in the clinic are even less clear than those of MDR1 and MRP1.
Another important feature of MDR1 and MRP in vitro is that drug resistance may be reversed, in part, after treatment with various agents that have distinctly different structural and functional characteristics, such as the calcium-channel blockers, calmodulin inhibitors, detergents, progestational and antiestrogenic agents, antibiotics, antihypertensives, antiarrhythmics, antimalarials, and immunosuppressives.
130,
131,
132 These agents bind directly to Pgp, thereby blocking the efflux of the cytotoxic drugs and increasing intracellular drug concentrations. Therefore, the role of MDR modulators has been a source of great contemporary interest, but the interpretation of clinical studies of resistance modulation has been confounded by the fact that MDR modulators, particularly MDR1 reversal agents, also enhance drug uptake in normal cells, decrease biliary elimination and drug clearance, and lead to enhanced toxicity.
130,
131,
132 Overall, strategies aimed at reversing resistance to the vinca alkaloids in the clinic with pharmacologic modulators of both MDR1 and MRP1 have been disappointing, most likely due to the fact that many other proteins besides MDR1 and MRP1 occur in association with the MDR phenotype.
92 Nevertheless, by characterizing the genetics and role of the ABC transporters in normal organ function and in the disposition of chemotherapeutic agents, there is a great deal to learn about how genetic polymorphisms in these proteins impact pharmacokinetics and drug toxicity.
The second well-characterized mechanism of vinca alkaloid resistance relates to tubulin isotypes. Mammalian cells have six
α– and seven
β-tubulin isotypes, whose expression may influence microtubule dynamics. Structural alterations in
α– or
β-tubulin due to either genetic mutations and consequential amino acid substitutions or posttranslational modifications, particularly phosphorylation and acetylation, have been identified in cancer cells with acquired resistance to the vinca alkaloids.
18,
19,
57,
69,
117,
118,
133,
134,
135,
136,
137,
138,
139,
140,
141,
142 These alterations result in
α– and
β-tubulins that confer hyperstability to microtubule polymers and are collaterally sensitive to the taxanes and similar tubulin-stabilizing natural products (see “
Mechanisms of Resistance” under “
Taxanes”). Although the means by which tubulin alterations confer resistance to the vinca alkaloids are not entirely clear, this phenomenon is not apparently due to decreased binding affinity of the altered tubulins for drug.
138,
139,
140,
141 Instead, alterations in
α– and
β-tubulins promote resistance to agents that inhibit microtubule assembly by increasing microtubule stability, perhaps by promoting longitudinal interdimer and intradimer interactions and/or lateral interactions between protofilaments.
142Decreased expression of class III
β-tubulin, which increases the rate of microtubule assembly, in contrast to overexpression of class III
β-tubulin, which is associated with rapid disassembly, is associated vinca alkaloid resistance in vitro.
117,
118,
137,
142,
143,
144,
145,
146,
147 Additionally, knockdown of either class II or IVb
β-tubulin with siRNA hypersensitizes lung cancer cell lines to the effects of the vinca alkaloids, with the effects more pronounced following knockdown of class IVb
β tubulin.
118,
137,
147 A high level of class III
β-tubulin expression also appears to independently predict a poor response and reduced survival in patients with non-small cell lung and breast cancer, following treatment with VRL or the taxanes.
117,
118,
147,
148,
149,
150 These data suggest that class III
β-tubulin may alter the dynamic
behavior of microtubules, as well as their sensitivity to inhibitors, but the overexpression of both class III and IVa
β-tubulin appears to be more germane to conferring resistance to the taxanes and other microtubule-stabilizing agents (see the section on “
Mechanisms of Resistance” under “
Taxanes”). The expression of class II
β-tubulin, which appears to be linked to p53 suppressor function, may also relate to vinca alkaloid resistance.
117,
151Increased expression of MAPs, particularly MAP4, which promote microtubule assembly and hyperstability, perhaps by mechanisms similar to those linked with alterations in
α– and
β-tubulins, has also been associated with vinca alkaloid resistance.
152,
153Various alterations in the principal components of the apoptotic pathway may confer resistance to the vinca alkaloids. Although much more work has been done in this area with the taxanes and various other microtubule-stabilizing agents (see the section on “
Mechanisms of Resistance” under “
Taxanes”), VFL resistance has been associated with down-regulation of the antiapoptotic factor Bcl-2, but this potential mechanism appears to be a cell type-dependent phenomenon.
154 There is also experimental evidence suggesting that VBL and other antimicrotubule agents induce proapoptotic effects by phosphorylating the antiapoptotic factor Bcl-xL, thereby reducing the formation of Bax, whereas some VBL-resistance mutants that undergo reduced apoptosis have defects in BcL-xL phosphorylation.
117,
118,
155
Drug Interactions
Pharmacokinetic interactions between the vinca alkaloids and other drugs, particularly cancer therapeutics developed over the last two decades, have not been studied in detail. Methotrexate accumulation in tumor cells is enhanced in vitro by the presence of VCR or VBL, an effect mediated by a vinca alkaloid-induced blockade of drug efflux; however, the minimal concentrations of VCR required to achieve this effect in myeloblasts (0.1 μmol/L) are achieved only momentarily in the clinic, and even higher concentrations are needed to enhance MTX uptake in lymphoblasts.
215,
216,
217 Furthermore, the schedule of VCR followed by MTX has not demonstrated synergism in the L1210 murine leukemia cells.
218 Cytotoxic synergy is noted with the sequence of MTX followed by VCR, but this interaction is not likely due to enhanced MTX uptake. Thus, very little justification exists for routine use of VCR pretreatment in high-dose MTX protocols. The vinca alkaloids also inhibit the cellular influx of the epipodophyllotoxins in vitro, resulting in less cytotoxicity, but the clinical significance of this observation is unclear.
219 L-asparaginase may reduce the hepatic clearance of the vinca alkaloids, particularly VCR, which may result in increased toxicity. To minimize the possibility of this interaction, VCR should be given 12 to 24 hours before L-asparaginase. In a murine model, VCR prevented doxorubicin-induced cardiomyocyte death, possibly by retarding the onset of apoptosis in association with the delay of poly(ADP)ribose polymerase activation.
220Treatment with the vinca alkaloids has precipitated seizures associated with subtherapeutic plasma phenytoin concentrations, most likely due to induction of CYP3A.
221,
222 Reduced plasma phenytoin levels have been noted from 1 to 10 days after treatment with both VCR and VBL. Administration of the vinca alkaloids with erythromycin, clarithromycin, itraconazole, posaconazole, voriconazole, and other inhibitors of CYP3A may also lead to severe toxicity.
178,
179,
180,
181,
182,
183,
184,
185,
223,
224 Concomitantly administered drugs, such as pentobarbital and H
2-receptor antagonists, may also influence VCR clearance by modulating hepatic cytochrome P450 metabolism.
217 Another potential drug interaction may occur in patients who have Kaposi’s sarcoma related to acquired immunodeficiency syndrome and are receiving concurrent treatment with 3′ azido-3′-deoxythymidine (AZT) and the vinca alkaloids, as the vinca alkaloids may inhibit glucuronidation of AZT to its 5′-
O-glucuronide metabolite.
225 Lastly, the significant interindividual and intraindividual variability of VCR pharmacokinetics in children has been attributed to the variable induction of P450 metabolism by P450-inducing corticosteroids.
226