Antimetabolites: Nucleoside and Base Analogs
ANALOGS OF DNA PRECURSORS; GENERAL CONSIDERATIONS
The synthesis of new DNA is an essential step in the replication of normal and malignant cells. Accordingly, the four bases that comprise DNA (the pyrimidines: cytosine, thymine; and the purines: adenine, and guanine) have provided a rational target for synthesis of analogues that inhibit the function of DNA, including its replication. These bases become active substrates for DNA synthesis through the attachment of deoxyribose sugars to form a deoxynucleoside. Three phosphate molecules must then be attached to the 5′-OH position of the nucleoside’s sugar, forming a metabolically active deoxynucleotide. These synthetic reactions, which lead to formation of the triphosphates required for making DNA, occur within the cancer cell, as well as within normal proliferating tissues, such as bone marrow and epithelium.
Normal as well as tumor cells do not have to synthesize bases for DNA. They can take up certain bases (guanine and uracil) as well as nucleosides (deoxycytidine, thymidine, adenosine, guanosine) from the circulation. Alternatively, these bases or their nucleosides can be synthesized by tumor cells de novo, in a complex, multistep system of reactions. Many of the earliest effective anticancer agents were designed as analogs of these bases or nucleosides. These analogs are transported into cells and converted to active triphosphates by the same transporters and enzymes that activate physiologic bases and deoxynucleosides.
FLUOROPYRIMIDINES
5-Fluoro-uracil (5-FU) and its prodrug, capecitabine (4-pentoxycarbonyl- 5′-deoxy-5′-fluorocytidine), are central agents in the treatment of epithelial cancers. They have synergistic interaction with other cytotoxic agents, such as cisplatin or oxaliplatin, and with radiation therapy. As a component of adjuvant and anti-metastatic therapy, fluoropyrimidines have improved survival in patients with colorectal cancer (1).
 MECHANISM OF ACTION AND RESISTANCE
MECHANISM OF ACTION AND RESISTANCE
The first agent of this class, 5-FU (Figure 1-1), was synthesized in 1956 by Heidelberger, based on experiments that demonstrated the ability of tumor cells to salvage uracil for DNA synthesis. Later work showed that 5-FU is converted by multiple different routes to an active deoxynucleotide, FdUMP, a potent inhibitor of thymidylate synthase (TS), and thereby, DNA synthesis (Figure 1-1).
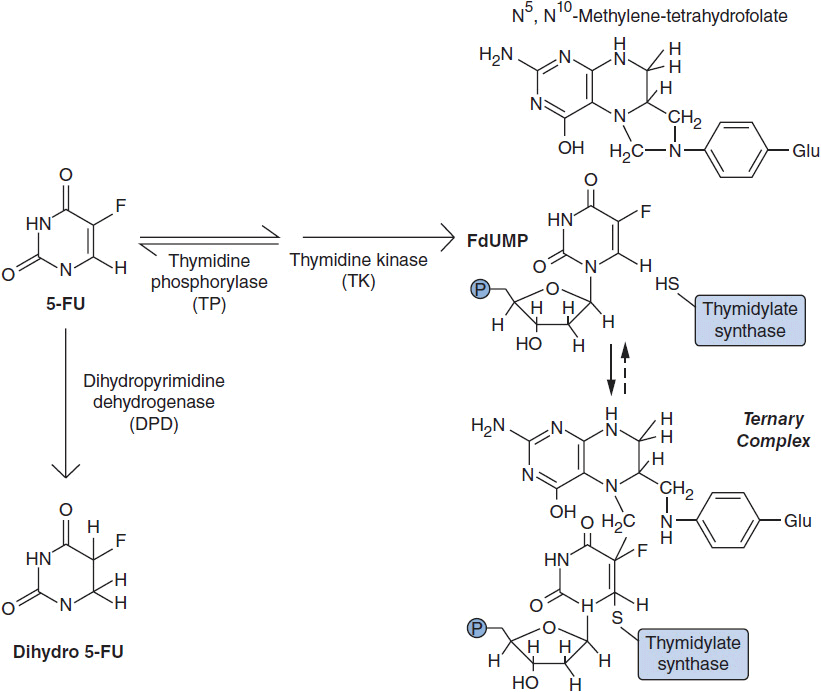
FIGURE 1-1 Routes of activation (via TP and TK) and inactivation (via DPD) of 5-fluorouracil (5-FU). Note that TP is a reversible reaction.
The active product, FdUMP, forms a tight tripartite complex with TS in the presence of the enzyme’s cofactor, 5-10-methylene tetrahydrofolic acid. It thereby blocks the conversion of dUMP to dTMP, a necessary precursor of dTTP (2). dTTP is one of four deoxynucleotide substrates required for synthesis of DNA. An exogenous folic acid source such as leucovorin (5-formyl-tetrahydrofolate) enhances formation of the TS-F-dUMP-folate complex and increases the response rate in patients with colon cancer (3).
5-FU also forms 5-FUTP, and becomes incorporated into RNA, where it blocks RNA processing and function. Inhibition of TS predominates as the mechanism of antitumor action.
Resistance to fluoropyrimidines arises through several mechanisms (4). Increased expression of TS, or amplification of the TS gene, occurs both experimentally and in a patient’s tumors after exposure to FU, and probably represents the primary mechanism. Some resistant tumors fail to convert 5-FU to its active nucleotide form through decreased expression of activating enzyme(s). Increased expression of degradative enzymes (thymidine phosphorylase [TP] and dihydropyrimidine dehydrogenase [DPD]); Figure 1-1 has been found in resistant cells. Increased expression of TP reduces the cellular pool of fluorodeoxyuridine, an intermediate in the activation pathway, and increases resistance. Upregulation of the AKT, RAS, and HER2 pathways may also contribute to resistance. Finally, anti-apoptotic changes, such as increased expression of bcl-2 or mutation of the cell cycle checkpoint, p53, are associated with resistance in experimental systems. A signature for resistance in patients treated with cisplatin and 5-FU for gastric cancer demonstrates increased expression of embryonic stem cell and PI-3-kinase pathway genes (5).
Capecitabine, an orally active prodrug of 5-FU, has demonstrated antitumor efficacy equal to 5-FU in breast and colon cancer. Capecitabine is activated by three sequential metabolic steps: (1) esterase cleavage of the aminoester at carbon 4 to yield fluoro-5′-deoxycytidine (F-5′-dC); (2) deamination of F-5′-dC, yielding fluoro-5′-deoxyuridine (ftorafur); and (3) cleavage of the inactive 5′-deoxy sugar of ftorafur by TP, releasing 5-FU (Figure 1-2). Steps 1 and 2 are believed to occur in the liver and plasma, while step 3, release of active 5-FU, takes place in tumor cells. Tumor cells with high TP are sensitive to capecitabine but resistant to 5-FU.
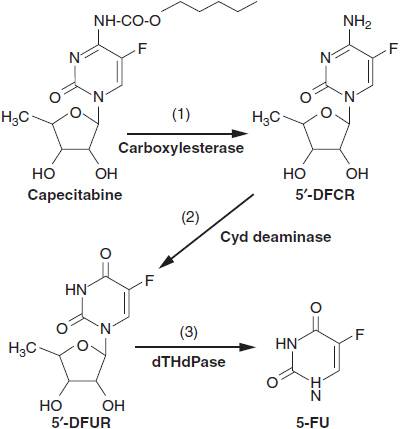
FIGURE 1-2 Metabolic activation of capecitabine by 1, carboxylesterase; 2, cytidine deaminase; 3, thymidine phosphorylase. 5-FU: 5-fluorouracil; 5′-DFCR: 5′-deoxy-5-fluoro-cytosine riboside; 5′-DFUR, 5′-deoxy-5-fluorouracil riboside.
Two other preparations of fluoropyrimidines, UFT and S-1, not available in the United States, incorporate ftorafur with inhibitors of DPD, yielding an orally active product that produces a long 5-fluorouracil half-life (2 h for S-1) in plasma, and somewhat increased epithelial toxicity (6). Both products are widely used (with leucovorin) in Japan but are not approved in the United States.
 CLINICAL PHARMACOLOGY
CLINICAL PHARMACOLOGY
5-FU is administered intravenously in several different regimens. It was originally given in doses up to 450 mg/m2/day × 5 days, and leucovorin, 25–500 mg/day orally, was later added to enhance efficacy. 5-FU given once weekly causes less neutropenia and diarrhea, and is probably equally effective. More recent and more effective regimens employ a bolus of FU on day 1, followed by 48-h infusion of up to 1000 mg/m2/day for 2 days. Bolus and infusion doses vary according to other drugs in the combination regimen and the use of radiation therapy concomitantly.
The parent drug is not readily bioavailable by the oral route due to rapid first-pass metabolism in the liver. Following intravenous administration, plasma concentrations of 5-FU decline rapidly, with a t1/2 of 10 min, due to the conversion of 5-FU to dihydro-5-FU by DPD. Intracellular concentrations of 5-FdUMP and other nucleotides build rapidly, and decay with a half-life of approximately 4 h. Little intact 5-FU appears in the urine. Drug doses do not have to be altered for abnormal hepatic or renal function.
Capecitabine, given in total doses of 2500 mg/m2/day for 14 days, is readily absorbed, converted to 5-fluoro-5′-deoxyuridine (5-F-5′-dU) by the liver, and peak levels of metabolites appear in plasma about 2 h after a dose. Food taken with capecitabine protects the drug from degradation and leads to higher active metabolite concentrations in plasma. 5-F-5′-dU, the primary active precursor of 5-FU, exits plasma with a t1/2 of 1 h. There is no evidence that leucovorin enhances the activity of capecitabine. Because the clearance of 5-F-5′-dU is delayed in patients with renal dysfunction, capecitabine should not be used in patients with severe renal failure (7). Patients with moderately impaired renal function (CCr of 30–50 ml/min) should receive 75% of a full dose.
In fluoropyrimidine therapy, doses should be adjusted according to white blood cell count, gastrointestinal symptoms, and cutaneous (palmar plantar dysesthesia) toxicity.
 TOXICITY
TOXICITY
Fluoropyrimidines cause significant acute toxicity to the gastrointestinal tract and bone marrow. Of primary concern are mucositis and diarrhea, which may lead to dehydration, sepsis, and death. The risk is greatest in the presence of myelosuppression. Persistent watery diarrhea should alert the patient to receive immediate medical attention. Women are more often affected than men, and elderly patients (above 70 years) are particularly vulnerable to 5-FU toxicity. Myelosuppression follows a typical pattern of an acute fall in white cell and platelet count over a 5–7 day period, followed by recovery by day 14. Occasional patients deficient in DPD due to inherited polymorphisms may display overwhelming toxicity to first doses of the drug (8). A test for DPD in white blood cells is now available, and can confirm this deficiency, which, if present, should preclude further attempts to use fluoropyrimidines. Other toxicities encountered with 5-FU include cardiac vasospasm with angina and rarely myocardial infarction and cerebellar dysfunction, the latter predominantly after high-dose intravenous or intracarotid infusion.
Capecitabine has the additional significant toxicity of palmar-plantar dysesthesias, with redness, extreme tenderness, and defoliation over the palms and plantar surfaces.
A third fluoropyrimidine, 5-F-deoxyuridine (5-F-dU), is used almost exclusively in regimens of hepatic artery infusion (0.3 mg/kg/day for 14 days) for metastases from colon cancer, in which setting it has a greater than 50% response rate (9). Given in this manner it has the advantage of achieving higher intratumoral concentrations. It is cleared by hepatic parenchyma and, by this route, produces modest systemic toxicity. Intrahepatic arterial infusion may lead to serious hepatobiliary toxicity, including cholestasis, hepatic enzyme elevations, and ultimately biliary sclerosis. Glucocorticoids given with 5-F-dU decrease the incidence of biliary toxicity. Thrombosis, hemorrhage or infection at the catheter site, and ulceration of the stomach or duodenum may further complicate this treatment approach.
CYTOSINE ARABINOSIDE
The first of these analogs, cytosine arabinoside (ara-C) (Figure 1-3), was isolated from a fungal broth and proved to be the single most effective drug for inducing remission in acute myelogenous leukemia (AML).
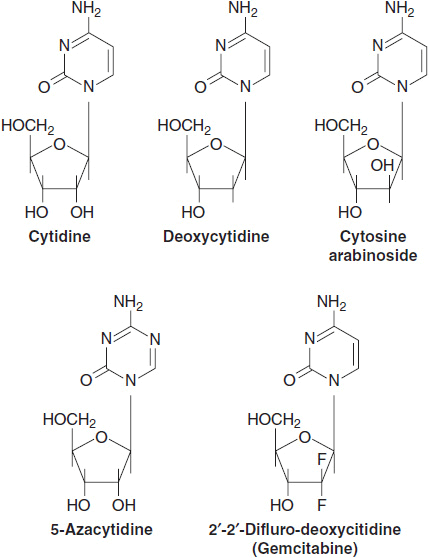
FIGURE 1-3 Structure of cytidine analogs.
It differs from the physiological substrate deoxycytidine in having an arabinose sugar rather than a deoxyribose, with a 2′-OH group in the abnormal beta configuration, rather than the 2′-H found on deoxyribose. The presence of the beta-2′-OH does not inhibit entry into cells or its further metabolism to an active triphosphate, or even its subsequent incorporation into the growing DNA strand. However, incorporation of a very few molecules of ara-C blocks further elongation of the DNA strand by DNA polymerase, and initiates apoptosis (programmed cell death) (10).
The steps leading from polymerase inhibition to cell death are not clearly understood. Exposure of cells to ara-C induces a complex set of reactive signals, including induction of the transcription factor AP-1, and the damage response factor, NF-κB. At low concentrations of ara-C, some leukemic cell lines in culture may differentiate, while others activate the apoptosis pathways. Exposure to ara-C leads to stalling of the replication fork for cells undergoing DNA synthesis, and this event activates checkpoint kinases, ATR and Chk 1, which block further cell cycle progression, activate DNA repair, and stabilize the replication fork. Loss of ATR or Chk 1 function sensitizes cells to ara-C. Levels of pro-apoptotic and anti-apoptotic factors within the leukemic cells also influence survival (11).
The specific steps in ara-C uptake and activation to a triphosphate within the cancer cell are important (Figure 1-4). It is taken into cells by an equilibrative cell membrane transporter, hENT1, which also transports physiologic nucleosides (12). Ara-C is then converted to its monophosphate by deoxycytidine kinase, a key rate-limiting step in its activation and antitumor action. Ara-CMP requires further conversion to its triphosphate, but the enzymes involved are found in abundance and do not limit its activity.
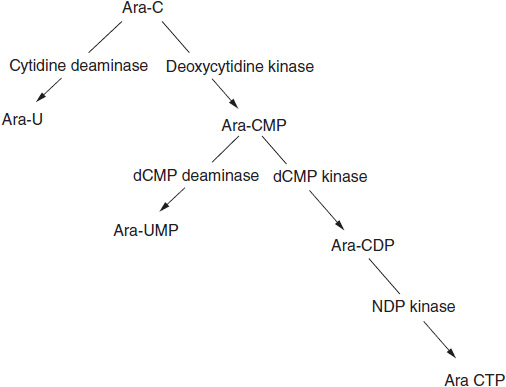
FIGURE 1-4 Metabolic pathway for conversion of deoxycytidine and its anticancer analog, cytosine arabinoside, to a triphosphate. Ara-CMP: ara-C monophosphate; ara-CDP: ara-C diphosphonate; ara-CTP: Ara-C triphosphate; ara-U: ara-uracil; dCMP: deoxycytidine monophosphate; NDP: nucleoside diphosphate.
The drug and its monophosphate, ara-CMP, are both subject to degradation by deamination. The resultant products, ara-U and ara-UMP, are inactive as a substrate for either RNA or DNA synthesis. Cytidine deaminase (CDA) is found in most human tissues, including epithelial cells of the intestine, the liver, and even in plasma. Elevated concentrations of CDA have been implicated as the cause of ara-C resistance in AML, but the evidence is as yet not convincing. Polymorphic variants of CDA (C-451T) decrease enzyme levels and are associated with greater toxicity and poorer survival (13). The most important cause of resistance appears to be a deletion of deoxycytidine kinase activity. Other evidence suggests that the pharmacokinetics (degree of formation and the duration of persistence) of ara-CTP in leukemic cells determine the therapeutic outcome. The intracellular half-life of ara-CTP is about 4 h. Exporters, particularly MRP 8, may reduce the intracellular drug levels and promote resistance (14).
High-dose ara-C has become the standard for consolidation of remission in AML, following remission induction. Cure rates for patients under 60 years of age now approach 30%–40%, but vary with patient age and with cytogenetics, the poorest results coming in older patients who have leukemia with complex karyotypes, leukemia secondary to cytotoxic therapy, or leukemia following a period of myelodysplasia.
 CLINICAL PHARMACOLOGY
CLINICAL PHARMACOLOGY
Ara-C, in doses of 100–200 mg/m2/day × 7 days, by continuous infusion, is commonly used with a topoisomerase 2 inhibitor (daunomycin or idarubicin) for remission induction in AML. Once remission has been induced, high-dose ara-C is given in doses of 1–3 g/m2 for consolidation therapy (15). Doses are repeated every 12 h twice daily on days 1, 3, and 5 in a commonly used schedule. Continuous infusion regimens are designed to maintain cytotoxic levels (above 0.1 µM) of drug throughout a several-day period, in order to expose dividing cells during the DNA synthetic phase of the cell cycle.
Ara-C disappears rapidly from plasma, with a half-life of 10 min, due primarily to its rapid deamination by CDA (see above). High-dose ara-C follows similar kinetics in plasma, although a slow terminal phase of disappearance becomes apparent, and may contribute to toxicity. The primary metabolite, ara-U, has no known toxicity, but, in patients with renal dysfunction, through feedback inhibition of deamination ara-U, may contribute to the slower elimination of high-dose ara-C from plasma, resulting in greater risk of toxicity. High-dose regimens provide cytotoxic drug concentrations in the cerebrospinal fluid, but direct intrathecal injection of 50 mg, either as a standard formulation of drug or in a depot form of ara-C immersed in a gel suspension for slow release (DepoCyt), is the preferred treatment for lymphomatous or carcinomatous meningeal disease. Ara-C has comparable intrathecal activity to methotrexate in these settings. DepoCyt produces sustained CSF concentrations of ara-C above 0.4 µM for 12–14 days, thus avoiding the need for more frequent lumbar punctures (16).
 TOXICITY
TOXICITY
Ara-C primarily affects dividing tissues such as the intestinal epithelium and bone marrow progenitors, leading to stomatitis, diarrhea, and myelosuppression, all of which peak at 7–14 days after treatment. In addition, ara-C may cause pulmonary vascular/epithelial injury, leading to a syndrome of non-cardiogenic pulmonary edema. Liver function abnormalities and rarely jaundice may occur as well, and are reversible with discontinuation of therapy.
High-dose ara-C may cause cerebellar dysfunction, seizures, dementia, and coma; this neurotoxicity is most common in patients with renal dysfunction and those over 60 years, thus leading to recommendations that highest dose consolidation (3 g/m2) not be used in such patients. The same neurotoxicities, as well as arachnoiditis, may follow intrathecal drug injection.
GEMCITABINE
A second deoxycytidine analog, gemcitabine (2′-2′-difluoro-deoxycytidine, dFdC, GEM), has become an important component of treatment regimens for pancreatic cancer, non-small cell lung cancer, and other solid tumors. Its metabolic pathways are similar to those of ara-C (Figure 1–4), although its triphosphate has a much longer intracellular half-life, perhaps accounting for its solid tumor activity. In vitro, sensitive tumor cells are killed by exposure to GEM concentration of 0.01 µM for 1 h or longer, levels achieved by usual intravenous doses.
Gemcitabine uptake and activation in tumor cells mimic that of ara-C, requiring the hENT1 transporter, initial phosphorylation to dFdCMP by deoxycytidine kinase (dCK), conversion to the triphosphate, and incorporation into DNA. It has additional sites of action. Its diphosphate forms an inhibitory complex with ribonucleotide reductase (RNR) (17), and thereby it lowers intracellular levels of its physiologic competitor, dCTP, allowing greater incorporation of dFdC into DNA. Experimental findings suggest that RNR inhibition is an important contributor to the toxicity of GEM. Incorporation into DNA leads to chain termination and apoptosis. Exposure of cells to GEM activates the same ATR/Chk 1 kinases that block further cell cycle progression after ara-C treatment, but, in addition, it activates ATM, a checkpoint pathway that responds to double-strand breaks, and thus its action may differ from the single break pathway activation by ara-C.
Resistance in experimental tumors arises by several mechanisms, including deletion of the hENT1 transporter, deletion of dCK, increased phosphatase activity, or increased expression or amplification of either the large, catalytic subunit of RNR or its smaller tyrosyl-containing subunit. In clinical studies, higher dCK activity may predict improved survival in pancreatic cancer patients treated with GEM (18).
 CLINICAL PHARMACOLOGY
CLINICAL PHARMACOLOGY
The standard regimen of administration is 1000 mg/m2 given as a 30-min infusion on days 1, 8, and 15 of a 28-day cycle. More prolonged periods of infusion, up to 150 min, may produce higher intracellular levels of dFdCTP, but also greater toxicity, and perhaps greater antitumor effects. Comparative trials of short- and long-infusion strategies are ongoing.
Doses may be modified for myelosuppression. GEM markedly sensitizes both normal and tumor tissues to concurrent radiation therapy, thus requiring drug dose reductions of 70%–80%. The mechanism of radiosensitization appears to be related to inhibition of repair of double-strand breaks and to inhibition of cell cycle progression. The drug is cleared rapidly from plasma by the ubiquitous cytoplasmic enzyme, CDA, and has a half-life of 15–20 min. Patients with the CDA variant, 27A>C, have delayed drug clearance and improved survival (19). Women and elderly patients may clear the drug more slowly, and all patients should be watched carefully for extreme myelosuppression.
 TOXICITY
TOXICITY
The primary toxicity of GEM is myelosuppression, which peaks in the third week of a 4-week schedule, blood counts usually recovering rapidly thereafter. Mild liver enzyme abnormalities may appear with longer term use. Pulmonary toxicity, with dyspnea and interstitial infiltrates, may occur in up to a quarter of patients treated with multiple cycles of the drug. In addition, patients on repeated cycles of GEM experience progressive anemia, which appears to have several components, including the direct effects of drug on red cell production, and the induction of hemolysis. After multiple cycles of treatment, a small but significant fraction of patients will experience a hemolytic-uremic syndrome (HUS), including anemia, edema and effusions, and a rising BUN (20). The HUS reverses with drug discontinuation, but in patients with pancreatic cancer, there may be no alternative effective therapy, and careful reinstitution of GEM at lower doses may be tried.
Severe toxicity has been reported in Japanese patients with an inactivating polymorphism of the CDA gene at position 208 was found (21), a variant associated with a fivefold slower clearance of the parent drug, as compared to nontoxic controls.
5-AZACYTIDINE (5AZAC)
5-Azacytidine (5azaC) (Figure 1–3) is both a cytotoxic and a differentiating agent, and has become a standard drug for treatment of myelodysplasia (22). Decitabine (DazaC), the closely related deoxy analog of 5azaC, is also approved for treatment of MDS and has the same mechanism of action. In MDS, which is characterized by refractory cytopenias and diverse chromosomal abnormalities, 5azaC reduces blood transfusion requirements and improves the platelet and mature granulocyte count in one-quarter to one-third of patients. While both analogs inhibit DNA synthesis and cause myelosuppression, their favorable effects on MDS are believed to be due to inhibition of DNA methyltransferase (DMT) and thereby activation of genes that induce maturation of hematopoietic cells.
5azaC and DazaC are transported into cells by nucleoside transporters, and are then converted to a nucleoside monophosphate by cytidine or deoxycytidine kinase, respectively. After further conversion to a triphosphate, they become incorporated into DNA, and act as a suicide inhibitor of the DMT, inducing expression of silenced genes (23). Thus, in non-cytotoxic concentrations in tissue culture, both analogs promote differentiation of both normal and malignant cells. In patients with sickle-cell anemia, 5azaC induces synthesis of hemoglobin F and thereby reduces the frequency of sickle-cell crisis and acute chest syndrome. However, DNA synthesis inhibitors, such as hydroxyurea (HU), have a similar effect on patients with sickle-cell anemia; thus it is unclear whether 5azaC’s beneficial effects are mediated by DNA demethylation or by inhibition of DNA synthesis (24).
The mechanism of 5azaC action in MDS likely relates to gene demethylation and induction of differentiation. Both global DNA demethylation and induction of specific genes follow azaC and DazaC treatment. An unfavorable response to DazaC correlates with an unfavourable (high) ratio of CDA to dCK activity, and to a lack of demethylation of selected genes (25). The pretreatment level of global methylation does not predict response.
 CLINICAL PHARMACOLOGY
CLINICAL PHARMACOLOGY
The elimination of both aza analogs occurs through their rapid deamination in plasma, liver, and other tissues by CDA. The plasma half-life of parent drugs is brief, 20–25 min. The primary metabolites, 5-azauridine and 5-deaza-deoxyuridine, undergo spontaneous hydrolysis and are inactive.
Toxicity of both aza analogs is primarily myelosuppression, with recovery 10–14 days after treatment. 5azaC causes significant nausea and vomiting when administered in high doses as antileukemic therapy. In occasional patients, hepatic dysfunction, rash, fever, or myalgias may be reported. In the usual regimen for myelodysplasia, 5azaC doses of 75 mg/m2/day × 7 days are repeated every 4 weeks. The drug has minimal side effects aside from leukopenia.
DazaC has more potent cytotoxic and differentiating properties, and causes leukopenia and thrombocytopenia as its major toxicities. Doses of 10 mg/m2/day × 5 days, in some regimens repeated on days 8–12, as tolerated, are given every 4 weeks in MDS treatment, but may induce prolonged neutropenia in patients with low WBC counts (25).
HYDROXYUREA
Hydroxyurea (HU), an inhibitor of RNR (26), is a useful agent for acutely lowering the white blood cell count in patients with myeloproliferative disease, especially acute or chronic myelogenous leukemia (CML). It also effectively lowers the platelet count in essential thrombocythemia. It has little value as a remission-inducing agent. Prior to imatinib, HU was a component of the maintenance regimen for CML but is now rarely employed for that purpose. Its effects on myelopoiesis are seen within 24 h, and reverse rapidly thereafter. Because of its minimal side effects and predictable and reversible action, it is commonly used to lower high white blood cell counts at the time of initial presentation of leukemia. It is also a potent radiosensitizer, and has been used with radiation therapy in experimental protocols for treatment of cervical cancer and head and neck cancer. It strongly induces fetal hemoglobin expression, and has become the standard agent for prevention of sickle-cell crisis (27). It has multiple effects on sickling, including an induction of fetal Hb, a reduction of adhesion of red cells to vascular endothelium, and a lowering of the white cell count, all of which may contribute to its beneficial action.
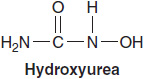
HU inhibits RNR by binding to the iron required for catalytic reduction of nucleoside diphosphates. Through deoxynucleotide depletion, it blocks progression of cells through the DNA synthetic phase of the cell cycle. Through its effects on deoxynucleotide pools, it enhances incorporation of other antimetabolites into DNA, and inhibits repair of alkylation. Resistance arises through outgrowth of cells that amplify or overexpress the catalytic subunit of RNR.
In addition to its effects on DNA synthesis, HU stimulates production of nitric oxide by neutrophils; NO in turn may function as an inducer of differentiation and a vasodilator, effects that may contribute to its control of sickle-cell crisis.
 CLINICAL PHARMACOLOGY
CLINICAL PHARMACOLOGY
HU is well absorbed after oral administration, but is available for intravenous infusion as well for emergent situations. Usual daily oral doses are 15–30 mg/kg, although higher doses are used for acute lowering of the white cell count. It is cleared by renal excretion, and its plasma half-life is approximately 4 h in patients with normal renal function. Doses should be adjusted according to creatinine clearance in patients with abnormal renal function.
Its toxicity is manifest primarily as acute myelosuppression, affecting all three lineages of blood cells. It may also cause a mild chronic gastritis, an interstitial pneumonitis, skin hyperpigmentation, ulcerations on the lower extremities, and neurologic dysfunction. It is a potent teratogen and should not be used without contraception in women of childbearing age. It has uncertain potential as a carcinogen, a concern in patients with nonmalignant disease and in chronic myeloproliferative syndromes such as polycythemia vera.
PURINE ANTAGONISTS
At least three general classes of purine antagonists have proven useful for treatment of cancer. The first were the thiopurines, 6-mercaptopurine (6-MP), and 6-thioguanine (6-TG), which were introduced as antileukemic drugs in the early 1950s (Figure 1–5). 6-MP remains a standard drug for maintenance of remission in childhood acute lymphocytic leukemia, in combination with methotrexate. 6-MP, the active metabolite of Imuran, is a potent immunosuppressive agent and is commonly used for Crohn’s disease. The second group (Figure 1–6) of purine analogs consists of halogenated adenosine derivatives, fludarabine, clofarabine, and cladribine. Unlike adenosine, these drugs are resistant to deamination, and are toxic to both normal and malignant lymphoid cells. Cladribine is highly effective, and possibly curative for hairy cell leukemia, while fludarabine has become a first-line agent for chronic lymphocytic leukemia and for follicular lymphomas (28). Fludarabine suppresses T-cell function and is effective against graft versus host disease when used with low dose irradiation in allogeneic bone marrow transplantation. Finally, nelarabine, an arabinosyl guanine (ara-G), is specifically effective against T-cell lymphoid tumors (29).
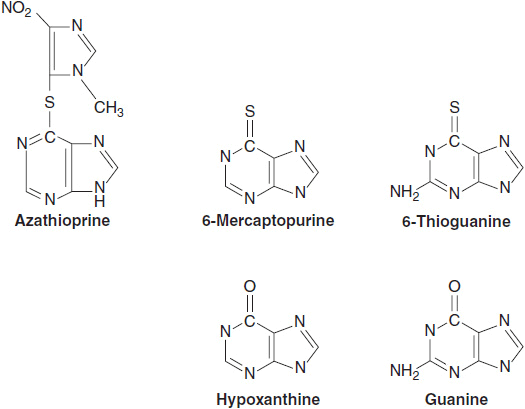
FIGURE 1–5 Structure of the naturally occurring purines, hypoxanthine and guanine, and related antineoplastic agents 6-mercaptopurine and 6-thioguanine, and the immunosupressive agent azathioprine.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


