Antihistamines
Jonathan A. Bernstein
Introduction
Histamine, a low-molecular amine, is produced by the reaction of histidine decarboxylase on L-histidine (1,2). This enzyme is located in cells found throughout the body including the central nervous system, gastric parietal cells, mast cells, and basophils (1,2). As a result of its ubiquitous presence in the body, it is not surprising that histamine has a wide range of biologic effects. Currently it is known that histamine exerts these effects through four histamine receptors (HRs). Histamine is involved in sleeping and waking, energy and endocrine homeostasis, cognition and memory through histamine receptor 1 (H1R), regulation of gastric acid secretion through H2R, modulation of neurotransmitter release through H3R, and facilitation of pro-inflammatory activities through H4R (Table 33.1) (1). HR antagonists (antihistamines) can be categorized in terms of their structure, pharmacokinetics, pharmacodynamics, and clinical utility (Table 33.1) (1). Histamine binding to H1Rs causes itching, pain, vasodilatation, vascular permeability, hypotension, flushing, headache, tachycardia, bronchoconstriction, stimulation of airway vagal afferent nerves and cough receptors, and decreased atrioventricular-node conduction time (1,2). Histamine binding to H2Rs causes increased gastric acid secretion, vascular permeability, hypotension, flushing, headache, tachycardia, chronotropic and inotropic activity, bronchodilation, and airway mucus production (1,2). Histamine binding to H3Rs prevents excessive bronchoconstriction and mediates pruritus through nonmast cell pathways (1,2). Finally, histamine binding to H4Rs is important for differentiation of myeloblasts and promyelocytes. Histamine has been shown to have a number of immunomodulatory effects through these various receptors. Clinically, selective antagonists are available for blocking H1 and H2 receptors (Table 33.1) (1). Nonselective H3– and H4-receptor antagonists are available as research tools but not for clinical use at the present time (Table 33.1) (1). Second-generation antihistamines, many of which have been derived from first-generation agents, are more selective for H1Rs and have added a new dimension to the treatment of allergic disorders. Over the last several years, additional second-generation antihistamines have been introduced to the market and others, still under investigation may become available in the near future. This chapter will provide an overview of the immunologic and clinical effects of histamine so that the reader can appreciate the evolving role of histamine-antagonists in a broad spectrum of clinical disorders.
Table 33.1 Characteristics of Histamine Receptors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The Discovery of Histamine and Histamine Receptors: A Historical Perspective
Histamine or β-imidazolylethylamine was first synthesized by Windaus and Vogt in 1907 (3). The term histamine was adopted because of its prevalence in animal and human tissues (hist: relating to tissue) and its amine structure (Figure 33.1) (4,5). Dale and Laidlaw (6) in 1910 were the first to report histamine’s role in anaphylaxis when they observed a dramatic bronchospastic and
vasodilatory effect in animals injected intravenously with this compound. Subsequently, histamine was found to be synthesized from L-histidine by L-histidine decarboxylase and metabolized by histamine N-methyltransferase to form N-methylhistidine or by diamine oxidase to form imidazole acetic acid (7). However, only the N-methyltransferase pathway is active in the central nervous system. Originally, histamine’s classic physiologic actions of bronchoconstriction and vasodilation were believed responsible for the symptoms of allergic diseases through its action at one type of HR. In 1966, Ash and Schild were the first to recognize that histamine-mediated reactions occurred through more than one receptor based on observations that histamine had an array of actions such as contraction of guinea pig ileal smooth muscle, inhibition of rat uterine contractions, and suppression of gastric acid secretion (8). This speculation was confirmed in 1972 by Black et al., who used the experimental histamine antagonists, mepyramine and burimamide, to block histamine-induced reactions in animals (9). They observed that each of these antagonists inhibited different physiologic responses, suggesting that there were at least two histamine receptors, now referred to as H1 and H2 (9). Arrang et al., discovered a third histamine receptor (H3) with unique physiologic properties, raising the possibility that additional, yet unrecognized, histamine receptors exist (10). Table 33.1 summarizes the pharmacodynamic effects after activation of the known histamine receptors and their common agonists and antagonists (5,10,11). Characterizing histamine receptors has been essential in discovering histamine’s physiologic actions on target cells which include increased mucus secretion, increased nitrous oxide formation, endothelial cell contraction leading to increased vascular permeability, gastric acid secretion, bronchial relaxation, and suppressor T-cell stimulation. Finally, the H4R was discovered based on a genomic approach using the H3R sequence.
As mentioned, this receptor is expressed more selectively on dendritic cells, mast cells, eosinophils, monocytes, basophils, and T cells; therefore, it is believed to have an important immunoregulatory role (12). Figure 33.1 illustrates the actions of histamine on histamine receptors in allergic inflammation (13).
vasodilatory effect in animals injected intravenously with this compound. Subsequently, histamine was found to be synthesized from L-histidine by L-histidine decarboxylase and metabolized by histamine N-methyltransferase to form N-methylhistidine or by diamine oxidase to form imidazole acetic acid (7). However, only the N-methyltransferase pathway is active in the central nervous system. Originally, histamine’s classic physiologic actions of bronchoconstriction and vasodilation were believed responsible for the symptoms of allergic diseases through its action at one type of HR. In 1966, Ash and Schild were the first to recognize that histamine-mediated reactions occurred through more than one receptor based on observations that histamine had an array of actions such as contraction of guinea pig ileal smooth muscle, inhibition of rat uterine contractions, and suppression of gastric acid secretion (8). This speculation was confirmed in 1972 by Black et al., who used the experimental histamine antagonists, mepyramine and burimamide, to block histamine-induced reactions in animals (9). They observed that each of these antagonists inhibited different physiologic responses, suggesting that there were at least two histamine receptors, now referred to as H1 and H2 (9). Arrang et al., discovered a third histamine receptor (H3) with unique physiologic properties, raising the possibility that additional, yet unrecognized, histamine receptors exist (10). Table 33.1 summarizes the pharmacodynamic effects after activation of the known histamine receptors and their common agonists and antagonists (5,10,11). Characterizing histamine receptors has been essential in discovering histamine’s physiologic actions on target cells which include increased mucus secretion, increased nitrous oxide formation, endothelial cell contraction leading to increased vascular permeability, gastric acid secretion, bronchial relaxation, and suppressor T-cell stimulation. Finally, the H4R was discovered based on a genomic approach using the H3R sequence.
As mentioned, this receptor is expressed more selectively on dendritic cells, mast cells, eosinophils, monocytes, basophils, and T cells; therefore, it is believed to have an important immunoregulatory role (12). Figure 33.1 illustrates the actions of histamine on histamine receptors in allergic inflammation (13).
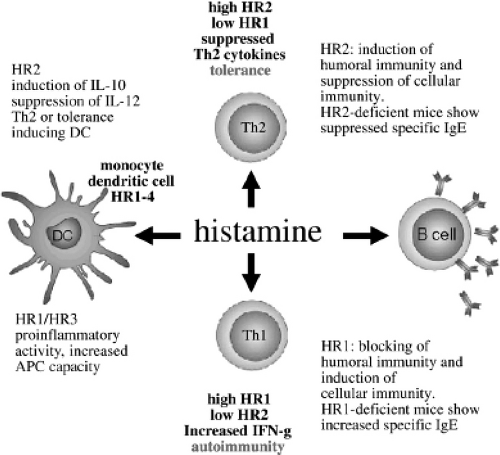 Figure 33.1 Function of histamine on histamine receptors. (Reproduced with permission from Simons FER, Simons KJ. The pharmacology and use of H1-receptor-antagonist drugs. NEJM 1994; 330:1663.) |
The Role of Histamine in Allergic Inflammation
Histamine is stored in the cytoplasm of mast cells and basophils, attached to anionic carboxylate and sulfate groups on secretory granules (13,14). Histamine is released from mast cell and basophil secretory granules after aggregation of high-affinity immunoglobulin E (IgE) receptors. IgE receptors are coupled to G-proteins which when activated lead to a sequence of chemical reactions with the end result being histamine release. However, histamine can be released spontaneously by activation of mast cells and basophils by histamine releasing factors, which include chemokines (RANTES, MCP-1, and MIP-1 α) and several cytokines (IL-1, IL-3, IL-5, IL-6, IL-7) (7).
Histamine’s inflammatory action depends on which histamine receptors are activated, the level of histamine receptor expression, and the effector cells involved (1,2). For example, H1R expression is increased during the differentiation of monocytes to macrophages and H1R expression can be increased by a number of inflammatory stimuli (1,2,12,13). Furthermore, histamine has varying effects on different inflammatory cells. For example, mast cells express H1, H2, and H4 receptors (1,2,12,13). Although histamine does not appear to have a direct effect on mast cell degranulation, by binding to H4Rs it can act synergistically with chemoattractants such as CXCL12 (1). In contrast, histamine binding to H2 receptors on mast cells can act to inhibit histamine release and modulate cytokine production (1,2,13).
Low concentrations of histamine via H4 receptors can induce eosinophil chemotaxis but at higher concentrations via H2 receptors can attenuate this effect (1,2,14). Histamine binding to H4 receptors can stimulate upregulation of adhesion molecules and reorganization of actin polymers, whereas binding to H1 receptors can induce superoxide production and complement receptor upregulation in eosinophils (1,2,12,13).
Dendritic cells express H1, H2, and H4 receptors (1,13). Histamine can cause chemotaxis of dendritic cells by binding primarily to H4Rs and to a lesser extent to H1Rs (1). Furthermore, T-cell polarization may be regulated by histamine binding via H1 and H4 receptors on dendritic cells in conjunction with other chemokines: CCL17, thymus and activation regulated chemokine (TARC); CCL22; CCL3 macrophage inflammatory protein 1 α (MIP1α).
Histamine can have direct effects on T cells via H1, H2, and H4 receptors which are expressed on CD4+ and CD8+ T cells (1). The effect of histamine on T-cell
proliferation varies (1). It can increase T-cell proliferation by binding to H1Rs and inhibit proliferation by binding to H2Rs. Binding to H2Rs has been demonstrated to inhibit T-cell production of IL-2, IL-4, IL-13, and interferon γ (1). However, the role of histamine in regulating T-cell proliferation is likely much more complicated than can be explained by the counter-regulatory roles of cytokine production (1).
proliferation varies (1). It can increase T-cell proliferation by binding to H1Rs and inhibit proliferation by binding to H2Rs. Binding to H2Rs has been demonstrated to inhibit T-cell production of IL-2, IL-4, IL-13, and interferon γ (1). However, the role of histamine in regulating T-cell proliferation is likely much more complicated than can be explained by the counter-regulatory roles of cytokine production (1).
H1-Receptor Histamine Antagonists
First-Generation Agents
Structure
The first histamine antagonist was accidentally discovered in 1937 by Bovet and Staub who found that a drug originally being studied for its adrenergic antagonistic properties in guinea pigs also had potent antihistaminic activity (5). By 1942, safe and effective antihistamines developed for human use became available. Many of these agents, such as pyrilamine maleate, tripelennamine, and diphenhydramine, are still widely prescribed today (2,5).
The chemical structure of H1-antagonists differs substantially from histamine (Figure 33.2) (1). Histamine is composed of a single imidazole heterocyclic ring linked to an ethylamine group, whereas H1 antagonists consist of one or two heterocyclic or aromatic rings joined to a “linkage atom” (nitrogen, oxygen, or carbon) (Table 33.2) (1,5). The linkage atom is important in structurally differentiating these groups of agents, whereas the number of alkyl substitutions and heterocyclic or aromatic rings determines their lipophilic nature
(1,5). The ethylenediamines, phenothiazines, piperazines, and piperidines all contain nitrogen as their linkage atom, whereas the ethanolamines contain oxygen and the alkylamines contain carbon as their linkage atoms (2,5).
(1,5). The ethylenediamines, phenothiazines, piperazines, and piperidines all contain nitrogen as their linkage atom, whereas the ethanolamines contain oxygen and the alkylamines contain carbon as their linkage atoms (2,5).
Table 33.2 Classification of Common H1-(First-Generation) Antagonists11 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Pharmacokinetics
Accurate pharmacokinetic data on first-generation antihistamines are now available in children and adults because of sensitive detection techniques, such as gas-liquid chromatography, mass spectrometry, and high-performance liquid chromatography (2,5,11). Generally, these compounds are rapidly absorbed orally or intravenously, resulting in peak serum concentrations within 2 to 3 hours and symptomatic relief within 30 minutes. They have large volumes of distribution, slow clearance rates, and are metabolized primarily by hydroxylation in the hepatic cytochrome P450 system. The vast majority of the parent drug is excreted as inactive metabolites in the urine within 24 hours of dosing. As a rule, serum half-lives (t1/2) are longer in adults than they are for children. Their lipophilic nature allows them to cross the placenta and the blood–brain barrier. This access into the central nervous system is responsible for many of the side effects experienced by patients. These agents are also excreted in breast milk (2,5,11). Table 33.3 summarizes pharmacokinetic and pharmacodynamic data for the most commonly used first- and second-generation agents (2,5,14).
Table 33.3 Pharmaccokinetics and Pharmacodynamics of Oral H1-antagonists in Healthy Young Adults | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||