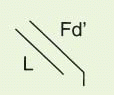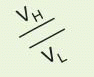Monoclonal antibodies (mAbs) are remarkably versatile agents with potential therapeutic applications in a number of human diseases, including cancer. Ten mAbs are now Food and Drug Administration (FDA)-approved mAbs for the treatment of cancer. MAbs have long promised to offer a safe, specific approach to therapy. Preclinical evaluation and human clinical trials have identified new strategies for the use of mAbs, as well as a number of obstacles to their effective application. Although the use of antibodies as targeting agents dates to the 1950s,
1 clinical investigation of antibodies as a potential treatment for cancer could not be initiated until the late-1970s, when efficient and reliable methods for the production of mAbs were developed.
2Five approaches to mAb therapy are used in humans. First, mAbs can be used to focus an inflammatory response against a target cell. The binding of a mAb to a target cell can result in fixation of complement, which yields cell lysis or results in opsonization that marks the cell for lysis by various effector cells, such as natural killer (NK) cells, neutrophils, and monocytes. Second, mAbs may be used as carriers to deliver another small molecule, atom, radionuclide, peptide, or protein to a specific site in vivo. Third, mAbs may be directed at critical hormones, growth factors, interleukins, or other regulatory molecules or their receptors to control growth or other cell functions. Fourth, anti-idiotypic mAbs may be used as vaccines to generate an active immune response. Finally, mAbs may be used to speed the clearance of other drugs or toxins or fundamentally alter the pharmacokinetic properties of other therapeutic agents. For example, mAbs may be fused to drugs or factors to increase their plasma half-life, change their biodistribution, or render them multivalent. Alternatively, mAbs may be used to clear previously infused mAbs from the circulation.
Despite the diversity of approaches, significant problems remain that are peculiar to mAbs. MAbs are large, immunogenic proteins, often of rodent origin, that rapidly generate neutralizing responses in patients within days or weeks after their first injection. The sheer size of mAbs—150 kDa for immunoglobulin G to 950 kDa for immunoglobulin M, 100 times larger than that of typical drugs—makes their pharmacology (particularly diffusion into bulky tumors or other extravascular areas) problematic for effective use. Many early mAbs or mAb constructs were either poorly cytotoxic or relatively nonselective, which rendered them ineffective. Moreover, the high degree of mAb specificity that is routinely achievable may allow tumor cells that do not bear the specific antigen target to escape from cytotoxic effects. Tumor cells can also shed or modify the target to avoid mAb binding. Nonetheless, mAbs still have great potential to be safe and effective anticancer agents. Several areas have been defined in which mAbs can be effective, either alone or in combination with other, more conventional agents.
This chapter reviews the basic biochemical and biologic properties of mAbs and their most commonly used derivatives (immunotoxins, radioimmunoconjugates, mAb fragments), discusses the pharmacologic issues peculiar to mAbs, and outlines some of the important clinical results obtained with mAbs. Because mAbs and conjugates of mAbs represent many different drugs with characteristics that result from their origin (rodent or human), their isotypes, their structure, or the various conjugated toxic agents, generalizations about the properties of mAbs may not be possible. Treatment of cancer with mAbs is a rapidly changing field, and readers are encouraged to consult other reviews for more comprehensive discussions of individual areas.
3
Immunoglobulin Structure
Immunoglobulins are separated into five classes or isotypes based on structure and biologic properties: immunoglobulin M (IgM), immunoglobulin D (IgD), immunoglobulin E (IgE), immunoglobulin A (IgA), and immunoglobulin G (IgG). IgM is the primordial antibody whose expression by the B cell on its surface represents the commitment of that cell to a particular but broad recognition space that subsequently narrows as part of the maturation response induced by antigen interactions.
4 In some cases, the antibodies interact with specialized receptors that link their action to host cellular defenses; in others, the antibodies interact with the humoral complement system. IgG is further divided into four subclasses and IgA into two subclasses. Heritable deficiencies in individual immunoglobulin classes or IgG subclasses are associated with susceptibility to particular infections and autoimmune disorders.
5 Table 25-1 summarizes various features of the antibodies discussed in this section.
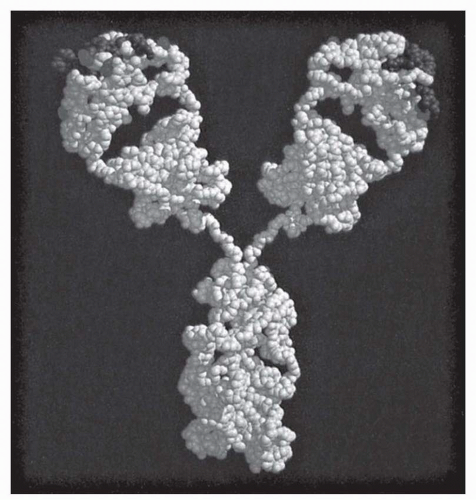
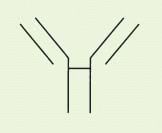
The fundamental structural elements of all antibodies are indicated by size as heavy and light chains of 55 to 75 kDa and 22 kDa, respectively (
Fig. 25-1). Light chains are either
κ or
λ and are each distributed among all immunoglobulin subclasses. Heavy chains are
μ, δ, γ, ε, and
α, corresponding to IgM, IgD, IgG, IgE, and IgA. The heavy chain isotype plays a role in defining the biologic characteristics of each antibody class. The amino-terminal domain of each chain is the variable (V
H or V
L) region that mediates antigen recognition; the remaining domains are constant regions designated C
L for light chain and C
H 1, C
H 2, and C
H 3 for heavy chain (and C
H 4 for
μ and
ε). Between C
H 1 and C
H 2 is the hinge region, which
confers flexibility on the antibody “arms” and susceptibility to proteases (see later), except in IgM and IgE, in which the C
H 2 domain itself serves this role.
Heavy (H) and light (L) chains are normally paired 1:1 with each other, but the smallest stable unit is a four-chain (HL)
2 structure (
Fig. 25-1), for a nominal total mass of 150 to 160 kDa for IgG and higher for other isotypes (
Table 25-1). IgE and IgG are composed of a single (HL)
2 unit, whereas IgM exists as a pentamer of (HL)
2 units joined by disulfide bonding with a third J-chain component. IgA exists mainly as a monomer in serum, but in secretions, it exists primarily as a dimer plus trimer and higher forms in which the oligomers are linked by J chain as well as the fragment of secretory chain (secretory piece) that is involved in the mucosal transport.
The V region itself is composed of subdomains—relatively conserved framework regions interdigitated with the complementarity-determining regions (CDRs; also termed “hypervariable segments” [HVSs]) that make primary contact with antigen (
Fig. 25-1).
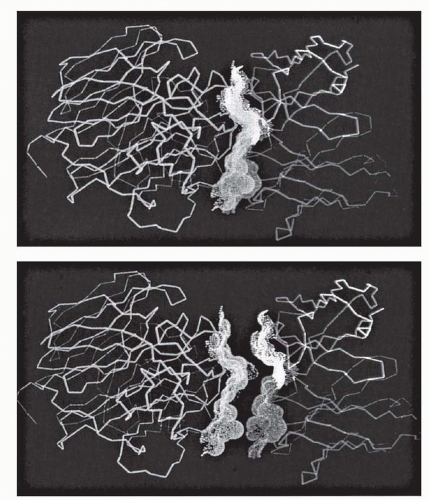
6 Three CDRs are found in each heavy and light chain that may participate in antigen binding. The V regions should be seen as juxtaposed three-fingered gloves, with the CDRs covering the tips (
Fig. 25-2), arrayed in a broad contact surface with antigen (
Fig. 25-3).
Antibodies are glycoproteins. Glycosylation of proteins plays various roles related to solubility, transport, conformation, function, and stability. Carbohydrate is located mainly in C domains, with a lower frequency in V regions (see data on M195 later).
7 IgG contains a major conserved glycosylation site in C
H 2 that contributes to the conformation of this domain, which is crucial to the functional ability to bind to complement and to Fc
γ receptors.
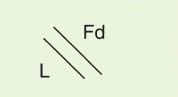
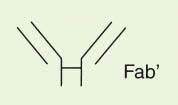
The IgG antibody “unit” has been defined in terms of susceptibility to proteases that cleave in the exposed, nonfolded regions of the antibody (
Fig. 25-1). A summary of antibody fragments and engineered or synthetic products is presented in
Table 25-2. Fab contains the V region and first C domain of the heavy chain (V
H + C
H 1 = Fd) and the entire light chain (L). Fab′ includes, in addition, a portion of the H chain hinge region and one or more free cysteines (Fd′). Fab′2 is a dimer of Fab′ linked through hinge disulfide(s). Fv is a semistable antibody fragment that includes only V
H + V
L, the smallest antigen-binding unit. Fc is the C-terminal crystallizable fragment that includes the complement and Fc receptor-binding domains (see below). Genetically engineered products include the
δ constructs; these lack the second C domain of heavy chain and behave like Fab′2, with bivalence, abbreviated survival and lack of interaction with host effector systems, but they do not require enzymic processing.
8 Another genetically engineered product, sFv (single-chain Fv), is Fv with a peptide linkage engineered to join the C-terminus of one chain to the N-terminus of the other for improved stability. More advanced products have been designed that conceptually represent the antigen-binding domain in a single peptide product
9; this is not related structurally to an antibody and is therefore considered an antibody mimic.
Ontogeny
Antibodies are a striking example of adaptation to the environment. This system allows the host to make molecules that bind antigens that the host has never encountered. The most diverse representation of classes and functions is found in Mammalia. The power of antigen recognition begins with an inherited array of duplicated and diversified germ-line V genes, a random mutational process that creates novel CDRs, a combinatorial selection process that amplifies the germ-line capabilities, and a controlled and directed mutational process that hones the specificity and matures the antibody into a high-affinity, antigen-specific reagent.
The biologic expression of antibody begins with the B-cell progenitor, which undergoes a series of maturation steps that begin with V gene selection for heavy chain followed by light chain V selection that yields surface expression and secretion by the mature B cell. On interaction with antigen, the B cells are activated
to proliferate, secrete antibody, undergo CDR mutagenesis and affinity maturation, and finally undergo chain switch and plasma cell differentiation. Plasma cells remain in tissues, spleen, or lymph nodes and secrete large quantities of antibody, which is the main function of this terminally differentiated cell.
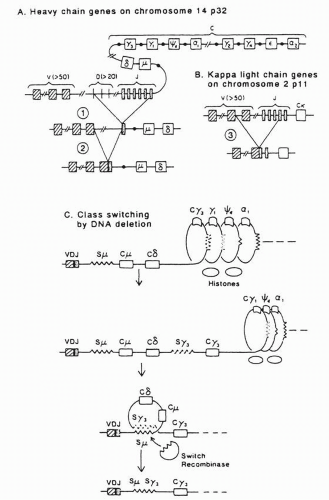
4The genes of heavy and light chains share important features of structure and maturation. Each gene locus contains widely separated V, C, and minigene domains that are placed into juxtaposition by DNA recombination mechanisms. The minigenes— diversity (D) and joining (J) regions for heavy chain and J regions for light chain—contribute to or constitute, with modifications, the CDR3.
10 The
κ and
λ light chain loci are located on chromosomes 2 and 22, respectively, but all heavy chains are contained within a single massive locus on chromosome 14.
Germ-line diversity is essential to the generation of the antibody repertoire. On the heavy chain locus are an estimated 80 functional V
H genes, 12 D regions, and 6 J regions for a potential of 6,000 combinations (
Fig. 25-4).
10,
11,
12 Roughly 80 V
κ light chain and 5 J
κ domains are found, which, randomly associated, can generate 400 combinations (the
λ locus contains a smaller number of distinct V genes). A simple arithmetical calculation suggests that V
κ V
H combinations alone could generate a diversity of approximately 2 × 10
6. Yet even this number is conservative because this diversity is amplified in turn by errors in recombination and processes called N and P nucleotide addition in CDR3, which add enormously to the potential complexity. This, in theory, exceeds the total lifetime B-cell output by several orders of magnitude.
10 Many authors, however, have cautioned that the mathematical diversity does not allow for the redundancy in configurations that could provide equivalent binding domains; in terms of antigen binding, the practical diversity is probably in range of 1 × 10
7.
V gene selection is based on random expression followed by specific amplification. The argument has been made on physicochemical grounds that 10
5 different antibody molecules are sufficient to create a topologic set that recognizes any antigen surface with an
affinity of 10
5 to 10
6 M
−1,
13 a weak but biologically important number that corresponds to recognition affinities of naive antibody-antigen contacts that are often broadly polyreactive. B cells express antibody, principally IgM and IgD, on their membranes. On contacting antigen, these cells are stimulated to divide and undergo CDR mutations. Subsequent binding and stimulation are in proportion to the strength of the binding reaction; hence, an in vivo selection occurs for mutations that enhance the affinity of the antibody for the antigen, a process termed “affinity maturation.”
14 Simultaneous with this increased affinity is a narrowing of the specificity, with the antibody shedding its early polyreactive phenotype. The cell then undergoes “class switch” to one of the mature antibodies (IgG, IgA, IgE) by deleting DNA between the VDJ region and the new C region of the heavy chain, which brings this new C domain in juxtaposition with the V region (
Fig. 25-4). (The light chain is unchanged.) Some time after commitment to a mature antibody, the cell ceases its CDR mutagenesis, affinity maturation is completed, and the B cell undergoes morphogenesis to a tissue-resident plasma cell.
4
Antigen-Antibody Interactions
Affinity is a quantitative measure of the strength of the interaction between an antibody and its cognate antigen, analogous to the equilibrium constant in the chemical mass action equation:

The equilibrium or affinity constant (Ka) is represented in units of M−1. In most instances studied by x-ray crystallography, contacts between antibody and protein antigen are dominated by noncovalent hydrogen bonds (O-H), with a lower frequency of salt bridges (COO− + H3N), for a total of 15 to 20 contacts. The effect of adding a new Hydrogen bond can be estimated from the free energy gain (0.5 to 1 kcal per mole) and from ΔG = −RT ln Ka to yield affinity increases of threefold to tenfold. Therefore, the affinity maturation that takes place (or affinity that may be lost in antibody engineering) changes quickly with a relatively small change in the number of bonds; that is, creating as few as three new hydrogen bonds may generate an affinity enhancement of more than 100-fold. This has been borne out by affinity changes that accompanied productive amino acid substitutions in V region engineering (see below). Of note, antibody affinities are generally much higher for protein antigens than for carbohydrate antigens, which may have less opportunity for hydrogen-bonding interactions (but are also “T-independent” antigens).
Although affinity and Ka directly express the binding potential of the antibody and are the most suitable measures for comparing affinities, the inverse of Ka, termed Kd or the dissociation constant, is expressed in molar units and indicates the concentration that is the middle of the range for the biologic action of the antibody:

That is, Kd is the concentration of free antibody at which antigen is 50% saturated; if the antibody is in large excess, the input antibody concentration approximates the free concentration. Kd is a frequently used term, but its relationship to affinity must always be borne in mind; that is, low affinity equals high Kd, and high affinity equals low Kd. For example, a Ka of 2 × 109 M−1 implies a Kd of 0.5 × 10-9 mol/L (0.5 nmol/L), or approximately 0.1 μg/mL antibody concentration for IgG. If antigen is in the picomolar (10−12 mol/L) range, this concentration of antibody will have half of the antigen saturated and half of the antigen will remain “free.” At 10-fold higher antibody concentration (1 μg/mL, 10 × Kd), antigen will be 90% saturated and 10% free, and at 10-fold higher concentration (10 μg/mL, 100 × Kd), antigen will be 99% saturated and only 1% unbound. A key point of understanding is that the ratio of antibody to antigen has very little impact on the degree of antigen saturation when antibody is in excess. If antibody concentration is 1 nmol/L with a Kd of 1 nmol/L, it does not matter whether antigen is 0.1 nmol/L at the Kd, 0.1 pmol/L, or 0.1 fmol/L; antigen in each case is 50% bound, although the ratio of antibody to antigen is 10, 104, and 107, respectively. Only the relation of free antibody to its Kd determines the degree of antigen saturation.
The affinity constant Ka is itself composed of two terms that describe the on-rate (forward; units of M−1 S−1) and off-rate (back; units of S−1) of the reaction:

To a first approximation, the forward rate is diffusion limited and is comparable for many antibodies reacting with macromolecules or cell-bound structures. Reactions of antibodies with haptens and other small molecules in solution are dominated by the faster linear and rotational diffusion rates of the smaller component.
15 For example, when 0.1 nmol/L of dinitrophenyl-lysine (0.1 ng/mL) or 0.1 nmol/L of cell-bound HLA-A2 (50 ng/mL) is mixed with specific IgG antibody at 10 μg/mL (65 nmol/L), 0.1 second is required for the antibody to react with 50% of the antigen for the hapten but 4 minutes is required to react with the surface protein. Yet, they have virtually the same affinity constant.15 This similarity is due to the fact that the fast association rate is balanced by a fast dissociation rate for the hapten (clearance half-time [
t1/2] = 0.7 seconds) whereas stability is longer for the protein antigen (
t1/2 = 6 minutes).
Although exceptions exist, the on-rates of antibodies to protein and cell-bound antigens are primarily in this range and inversely proportional to antibody concentration for antibody in excess of antigen (i.e., at 1 μg/mL, the 50% on time would be on the order of 0.5 to 1 hour). Accordingly, differences in affinity between antibodies to the same cellular antigen are in many instances reflective of the off-rate (
kb). For most purposes, an antibody is generally considered of “good” affinity if its
Ka is equal to or greater than 10
9 M
−1, where off-rate
t1/2 values of an hour or more at 4°C are common. Association and dissociation times at 37°C are both accelerated relative to the times at 4°C, on the order of 5 or more, frequently with a net decrease in antibody affinity of 2- to 10-fold. Temperature effects on affinity must be explicitly tested, however, because in some instances, protein-ligand affinities are enhanced by higher temperature.
16The foregoing expresses basic principles of binding processes. A further important feature of antibodies is their multivalent structures. Although the on-rates for monovalent Fab and bivalent Fab′2 constructs are comparable, the bivalent off-times may be 10-fold longer than for the monovalent constructs, which yield affinities that are similarly enhanced.
15 To discriminate the affinity that is intrinsic to the V region antigen interaction from the effective affinity in a bivalent or multivalent interaction, the latter is often referred to as
avidity. For monovalent interactions, avidity equals affinity; for multivalent interactions, avidity is greater than or equal to affinity. Theory predicts avidity enhancements that vastly exceed observed numbers, but structural constraints undoubtedly restrain the energy advantage of multivalent binding.
17,
18 In the extreme, steric factors constrain some bivalent antibodies (e.g., anti-Tac)
19 to bind only monovalently to antigens on cell surfaces although they will bind bivalently to antigen in solution. When antigens are presented multivalently on the surfaces of cells, viruses, or other pathogens, even the low-affinity IgM interactions can yield a high-avidity, stable binding to such targets in vivo.
Pharmacokinetics and Pharmacodynamics
The metabolism of immunoglobulins determines the duration of usefulness of antibodies in vivo. Under normal conditions, the serum levels of endogenous immunoglobulins are determined by a balance between synthetic and catabolic rates.
20 When antibodies are administered as therapeutics, these catabolic rates effectively specify the dose and schedule necessary to maintain therapeutic blood levels when steady-state exposures are targeted.
Table 25-1 lists the half-lives of human antibody survival in humans. IgG has the longest survival, 23 days (this value is for IgG1, IgG2, and IgG4; IgG3 survival is 7 days). A key element in the regulation of IgG catabolism is the Brambell receptor (FcRB), named after its discoverer, F.W.R. Brambell, who described this receptor more than 30 years ago (see Ref [
21] for a review).
21 This receptor is located in endosomes of endocytically active tissues, primarily vascular endothelium, which is mainly responsible for the catabolism of plasma proteins, including IgG. There, FcRB binds IgG, recycling it to the cell surface and diverting it from the pathway to lysosomes and the catabolism that is the fate of other, nonprotected proteins. In this role, FcRB is also termed the IgG protection receptor (FcRp). Yet FcRB is also responsible for transmission of IgG from mother to young, via yolk sac, placenta, or newborn intestine; in this manifestation, it is termed the IgG neonatal transport receptor (FcRn) for neonatal intestine, the tissue from which the receptor was initially cloned.
A substantial body of knowledge exists on the metabolism of immunoglobulins in various disease states. Conditions of protein wasting (enteropathies, vascular leak syndromes, burns), febrile states, hyperthyroidism, hypergammaglobulinemia, and inflammatory disorders are accompanied by significant acceleration of immunoglobulin catabolism.
20 This information is of importance for understanding in vivo survival data in various clinical applications. In fact, the controlled conditions of testing immunoglobulin metabolism are rarely duplicated in practice, with antibody survivals typically shorter than suggested by the numbers above. Typically, murine antibody survival
t1/2 values are in the range of less than 1 to 3 days, and antibodies with human gamma Fc domains (chimeric or humanized) have
t1/2 values in the range of 1 to 15 days. Some of this acceleration in clearance is clearly due to disease-associated catabolic factors and to antigen binding in vivo, but subtle changes in the drug structure during product preparation may have a role in this acceleration as well. The influence of antigen expression on antibody clearance in vivo is considered later.
Antibody fragments have been studied because of their abbreviated survival and because their small size may translate into better tissue penetration. Fab and Fab′2 have survivals in vivo of 2 to 5 hours in mice, with comparable values in humans, and survival is dependent largely on kidney filtration mechanisms.
20 Excretion is not based on size alone because the Fc fragment, which is comparable in size to Fab, is not filtered and has an in vivo survival of 10 days in humans. These rapidly catabolized fragments, like other filtered proteins, are largely absorbed in the proximal tubule and degraded to amino acids, which are returned to circulation. No intact immunoglobulin or fragments reenter circulation once filtered.
22 In normal kidney, less than 5% of filtered light chain is excreted intact, whereas this fraction increases in the setting of renal tubular disease.
23The effect of circulating antigen on the efficacy of antitumor therapies has been evaluated. This was first encountered in antiidiotype therapies directed at the surface Ig on B-cell lymphomas, some of which secreted high levels of idiotype.
24 Circulating idiotype prevented access of administered antibody to the idiotype on tumor cells, which effectively neutralized the drug, unless very high doses were given to overwhelm the secreted quantities of idiotype. Other targeted tumor antigens, including carcinoembryonic antigen (CEA),
25 gangliosides GD2 and GD3,
26,
27 and Tac,
28 also circulate at significant levels that might require adjustments to therapy.
Key observations include that there is (a) a direct relationship between the soluble antigen levels and the dose necessary to attain 50% (or 90% or 99%) bindability of administered antibody and (b) a predictable relationship between the rate of antigen synthesis and the time to antibody saturation.
28 The actual partitioning of antibody between soluble and cellular antigen depends on several features, including the effective avidity of antibody for antigen on cells (which may be higher than for soluble antigen) and other factors influencing tumor penetration.
A special concern in this setting is that many soluble forms of antigen have short
t1/2 values once shed from tumor cell surfaces, and the interaction with antibody may prolong their in vivo survival and increase their level in the whole body. When the target itself is a cytokine or cytokine receptor, adverse consequences conceivably could derive from the antibody treatment if the antigen retains activity in the antibody complex, as shown for interleukin-3, interleukin-4, and interleukin-7 complexes in vivo.
29 If the antigen does not retain activity in complex, then this problem causes no concern because the free concentration of antigen cannot be
increased by the presence of antibody, even after antibody is fully saturated with excess antigen. A different potential consequence of antigen load is that it may reduce transport of radioantibody to tumor for imaging or therapy. Studies have shown, however, that antibody can partition sufficiently between soluble and cellular antigen to yield targeting adequate for tumor-imaging purposes (CEA,
25 Tac
28).
Receptor Blockade
Antibody binding occurs without cooperation of other elements of the immune system. Therefore, just as antitoxin can prevent a toxin from acting at its target site in the body, antibody can also deny access of growth factors to tumors whose proliferation is factor dependent. This approach has been applied more widely in nonmalignant settings for the suppression of immune responses in autoimmune and alloimmune settings.
57 This approach is limited in malignancy because most tumors appear to be autonomous. In principle, an antibody directed against a cytokine should have the same result. However, the short half-lives of most cytokines and the locally high antibody concentrations required may make this approach more difficult. Such autocrine or paracrine loops may be better interrupted by an antireceptor than by an anticytokine antibody. One report has documented a marked
enhancement of cytokine activity by antibody to cytokine via
t1/2 prolongation, which runs counter to the goal of suppressing cytokine activity.
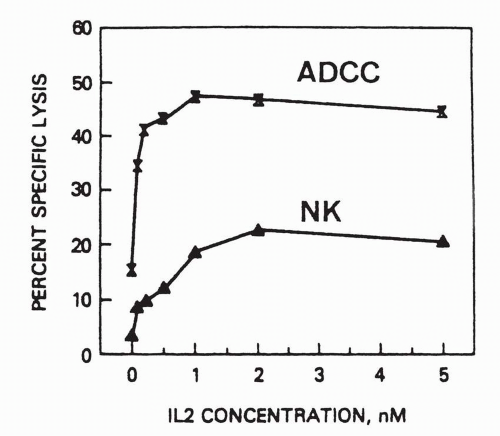
29Design of such applications also must consider the receptor occupancy that is necessary for cell survival and proliferation. Only 10% occupancy of the receptor for granulocyte-macrophage colony-stimulating factor is sufficient to induce maximal activation of granulocytes.
58 Similarly, one must block more than 90% of the
α chain of IL-2 receptor with antibody to have a significant impact on IL-2-dependent, antigen-induced T-cell proliferation.
47When the action of antibody is to attract effector cells, as with ADCC, then antigen expression correlates with target susceptibility. However, when the therapeutic action is to deprive a ligand access to its receptor, the level of expression of receptor antigen may have no bearing on the susceptibility of the target to antibody therapy. For example, the interleukin 2 receptor alpha chain (CD25)
confers high affinity binding of IL2 in conjunction the moderate affinity beta-gamma chain complex. On eosinophils, CD25 is present at levels that are below detection by flow cytometry or immunohistochemistry, yet antibody to the alpha chain is sufficient to ablate responsiveness of the eosinophils to interleukin-2 at physiologic concentrations.
59,
60 Similarly, much is made of the failure of the levels of epidermal growth factor receptor (EGFR) to predict responsiveness to anti-EGFR therapy, with “negative” tumors by immunohistochemistry being as responsive as positive tumor.
61,
62 This contrasts with another target of this family, Her2/neu, in which the responsiveness
is correlated with increased receptor expression.
63 The lesson here is that the receptor level that is detectable by standard methods is not necessarily reflective of the physiologic needs of the cell to respond to a receptor-ligand interaction, or predictive of the benefit to disrupting that interaction. Clinical testing of new antibodies should, therefore, not be restricted to patients whose tumors have been shown to express the target antigen.