Antiandrogen Therapy
The initial attempt at treating cancer with hormone ablation was implemented by Charles Huggins at the University of Chicago in 194l, when he hypothesized that the prostate depended on testosterone (and, ultimately, its metabolite dihydrotestosterone) for its growth (1). Orchiectomy of patients with advanced prostate cancer reduced serum testosterone to barely detectable levels (less than 50 ng/dl) and produced dramatic relief of bone pain in 90% of such patients. The median duration of response was about 1 year, and hormone-independent tumor emerged in most cases. Huggins was awarded a Nobel Prize for his work.
Since that time, androgen ablation has not changed in concept, but drugs have largely taken the place of orchiectomy. Three basic classes of drugs are used: (i) gonadotrophin releasing hormone (GnRH) agonists, a family of small peptides that promote release, and exhaustion of GnRH from the hypothalamus, thus lowering Gn levels in plasma and blocking androgen release from the testes (medical castration); (ii) small-molecular-weight androgen analogs that inhibit androgen interaction with its receptor in normal and tumor cells; and (iii) drugs that inhibit androgen synthesis in the adrenals and other peripheral, non-gonadal tissues (2).
GnRH AGONISTS
Two GnRH agonists approved for clinical use in the United States are leuprolide and goserelin. These drugs are given by intermittent, monthly to 4-monthly subcutaneous injection, and produce a flair response of testosterone release for several days to weeks, followed by a rapid decline in serum testosterone levels. The flair may induce an increase in bone pain, and in the presence of significant vertebral metastases, symptoms of spinal cord compression may result. In such patients a GnRH antagonist, abarelix, is available to ablate the flair response and offers protection from the short-term progression of disease (3), but is only occasionally used. A more important consideration in the use of GnRH agonists is their lack of effect on adrenal androgen, which makes a small contribution to serum androgen activity and theoretically could be sufficient to maintain or promote tumor growth in the absence of testicular androgen. However, clinical trials of complete androgen blockade with a GnRH agonist and an inhibitor of androgen receptor binding have not yielded conclusively positive results (4), as compared to GnRH agonists alone.
Side effects of GnRH agonists are those of acute androgen deprivation, including vasomotor instability (flushing and sweating), loss of libido, gynecomastia, acute and dramatic bone and muscle loss with an increase in hip fracture rates, truncal obesity, diabetes, and an increased risk of myocardial infarction and sudden cardiac death (5). A “metabolic syndrome” of insulin resistance, increased body fat mass, and changes in plasma lipids can be detected within weeks of initiation of GnRH therapy (6). Bone preservation with bisphosphonates is recommended for patients on long-term GnRH agonist therapy (7).
The GnRH analogs are cleared by both renal excretion and by hepatic metabolism, with an elimination half-life of 3–7 h. Depending on their formulation and dose, plasma concentrations of analog are sufficient to suppress testosterone levels for 1–4 months.
ANDROGEN RECEPTOR INHIBITORS
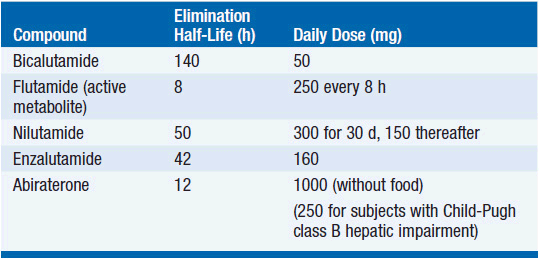
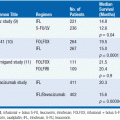
Four receptor antagonists and one androgen synthesis inhibitor (Table 12-1 and Figure 12-1) have been approved for treatment of prostate cancer. The older compounds (flutamide, bicalutamide, nilutamide) are commonly used with GnRH agonists to block the temporary surge in adrenogens released in response to GnRH agonists and to inhibit the residual effects of adrenal androgens. The newest receptor antagonist, enzalutamide is approved for treatment of advanced, castration resistant prostate cancer after progression on docetaxel (8). As single agents, flutamide, nilutamide, and bicalutamide are not as effective as the GnRH agonists, but their side effect profile is somewhat advantageous. They elevate testosterone levels as a result of inhibition of androgen receptors in the hypothalamus and increased GnRH secretion; thus as single agents they cause less loss of libido and gynecomastia, and have little effect on bone and muscle mass. However, all three drugs cause rare cases of severe hepatic injury, flutamide causes diarrhea, and nilutamide causes interstitial pneumonitis and visual disturbances (dark adaptation). All are eliminated by hepatic metabolism, and may inhibit the clearance of coumadin, phenytoin, and other agents cleared by hepatic cytochrome-dependent enzymes. Flutamide is rapidly converted to its active alpha-hydroxy metabolite after oral administration. The doses and pharmacokinetics of these antiandrogens are given in Table 12-1. Enzalutamide causes hot flashes, fatigue, and diarrhea as its major side effects; it causes seizures in suprapharmacological doses in animals, and this side effect has been reported in human subjects on this drug.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


