Chapter 1
- 1.
Protein hormones are stored within secretory vesicles and are secreted in response to a stimulus. Steroid hormones freely diffuse out of cells. Their synthesis, as opposed to secretion, is regulated by stimuli.
- 2.
Hormone-binding proteins in the serum generally increase the circulating half-life of a hormone. It is the free fraction (i.e., unbound) that is considered to be active.
- 3.
Increasing the GTPase activity of Gs would result in a more rapid inactivation of Gs, thereby decreasing adenylate cyclase activity and cAMP levels. Thus the cell would demonstrate resistance to hormones that act through Gs-coupled GPCRs.
- 4.
The IRS protein is recruited to and phosphorylated by the insulin receptor. The phosphotyrosines within the IRS protein recruit and activate the Ras-Raf-MAPK pathway, which transduces insulin receptor binding into a growth response. Other phosphotyrosines on the IRS protein activate the PI3 kinase–PKB pathway, which is linked primarily to metabolic actions of insulin (including GLUT4 translocation to the membrane).
- 5.
Cytokines and TGF-β–related hormones (e.g., inhibin) signal by phosphorylating a transcription factor (cytokines use STATs, and TGF-β–related hormones use SMADs). The transcription factors then translocate to the nucleus (as dimers) and activate the expression of specific genes.
- 6.
Hormone binding to a GPCR induces a conformational change in the receptor, which makes the receptor a substrate for serine/threonine GPCR kinases (GRKs). β-Arrestins then bind to the phosphorylated residue and link the receptor to clathrin-coated pits and clathrin-mediated endocytosis. Receptors within endosomes may recycle back to the membrane, or may be destroyed by lysosomal enzymes if the endosome fuses with a lysosome.
- 7.
Guanosine nucleotide exchange factors (GEFs) allow a small G protein to exchange a bound GDP (inactive state) for GTP (active state). GPCRs are essentially ligand-activated GEFs for the trimeric G-protein complexes. Without this activity, GPCRs could not activate the α subunits of the trimeric G protein complex.
- 8.
Phospholipase C (PLC) cleaves the membrane phospholipid, phosphatidylinositol, into diacylglycerol (DAG) and inositol-3-phosphate (IP 3 ). DAG activates certain isoforms of protein kinase C, whereas IP 3 binds to its receptor in the endoplasmic reticulum to induce release of Ca 2+ into the cytoplasm.
Chapter 2
- 1.
Cephalic, gastric, and intestinal. The greatest release of gastrin occurs during the gastric phase. This occurs because (1) the presence of amino acids in the stomach directly stimulates gastrin release from G cells; (2) the presence of food promotes gastrin release through pressor receptors in the stomach wall; (3) the presence of food buffers acidity, thereby decreasing inhibitory somatostatin release; and (4) there is minimal inhibitory signaling from the small intestine and colon.
- 2.
(a) Stimulation. (b) Stimulation. (c) Inhibition. (d) Stimulation. (e) Stimulation.
- 3.
S cells secrete secretin, I cells secrete CCK. CCK inhibits gastric motility. Secretin may act as an enterogastrone to inhibit gastrin secretion but has a minimal effect on gastric emptying.
- 4.
(a) Some stimulation. (b) Strong stimulation. (c) No effect. (d) Increase. (e) Decrease.
- 5.
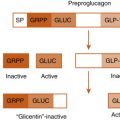
Both glucagon-like peptide-1 (GLP-1) and glucagon are encoded within the same gene that encodes the preproglucagon prohormone. Glucagon is secreted by α cells of the endocrine pancreas and binds to the glucagon receptor. GLP-1 is released by intestinal L cells and acts through the GLP-1 receptor.
- 6.
An incretin is a hormone (peptide) that is released in response to food (especially glucose) in the intestine and enhances the ability of glucose to promote insulin release from the pancreatic β cells. Two incretins are GIP and GLP-1.
- 7.
Hypertrophy and hyperplasia of the gastric mucosa and rugae (submucosal folds). Enterochromaffin-like (ECL) cells show the greatest degree of proliferation.
- 8.
Erythromycin binds to and activates the motilin receptor.
Chapter 3
- 1.
In the fed state, glycolysis in the liver leads to de novo fatty acid and triglyceride synthesis. In the adipose tissue, glycolysis generates glycerol-3-phosphate, which is used to reesterify fatty acids (from the digestion of chylomicrons by LPL) into triglycerides. Glycolysis is also used to generate ATP.
- 2.
Glycogen is a storage form of glucose. In the liver, glycogenolysis contributes directly to blood glucose because the liver can dephosphorylate glucose-6-phosphate to glucose. In the muscle, glycogen is used during exercise to provide ATP by glycolysis. Skeletal muscle cannot dephosphorylate glucose-6-phosphate and thus cannot directly contribute to blood glucose levels.
- 3.
Ketone bodies. The liver produces ketone bodies from free fatty acids and ketogenic amino acids.
- 4.
Accumulation of mitochondrial citrate (in times of plentiful ATP) can be transferred to the cytoplasm, where it can generate cytoplasmic acetyl CoA used for lipogenesis.
- 5.
Lipoprotein lipase (insulin-dependent activation) and hormone-sensitive lipase (insulin-dependent inhibition).
- 6.
LDL particles lose apoprotein E and bind to LDL receptors through apoprotein B100. LDL receptors remove these high cholesterol particles from the blood by receptor-mediated endocytosis. The liver plays the major role in LDL receptor–mediated removal of LDL particles, although steroidogenic cells and proliferating cells (i.e., cells that need cholesterol) also take up LDL particles by the LDL receptor. Loss of LDL receptor results in a high LDL concentration in the blood and, therefore, high cholesterol content in the blood.
- 7.
Malonyl CoA inhibits the carnitine-palmitoyl transferase-I transporter. This prevents the futile cycle of synthesizing fatty acids only to have them transported into the mitochondria for β-oxidation.
- 8.
Decreased glucokinase activity would inhibit glycolysis and therefore ATP production in β cells. Lower ATP would result in less insulin secreted. Heterozygous null mutations of glucokinase cause one form of MODY.
- 9.
- a.
Glucokinase—insulin increases gene expression through SREBP-1C;
- b.
Fructose-1,6-bisphosphatase—insulin represses gene expression through inactivation of FOXO1 and inhibits enzyme activity by increasing the level of fructose-2,6-bisphosphate, which is an allosteric inhibitor of fructose-1, 6-bisphosphatase.
- c.
Pyruvate kinase—insulin both increases PK gene expression through SREBBP-1C and increases PK activity through protein phosphatase-mediated dephosphorylation.
- d.
Acetyl CoA carboxylase—insulin increases ACC1 and ACC2 gene expression through SREBP-1C, and increases activity by phosphatase-mediated dephosphorylation.
- e.
PEPCK—insulin inhibits PEPCK gene expression, at least in part by inactivation of FOXO1.
- a.
- 10.
In the presence of a low I/G ratio, more fatty acids are released from adipose tissue. These fatty acids are transported to the liver where they are metabolized. β-Oxidation increases. The low I/G ratio inhibits glycolysis and lipogenesis. Thus malonyl CoA levels remain low. Malonyl CoA is an inhibitor of carnitine-palmitoyl transferase-I: while malonyl CoA levels drop, carnitine-palmitoyl transferase-I activity increases. Hepatocyte mitochondria contain the enzymes for β-oxidation and ketogenesis. The elevated acetyl CoA production from β-oxidation, along with the decreased TCA cycle activity caused by NAD depletion, results in increased ketone body production. T1DM also results in an increased hepatic glucose production; enhanced gluconeogenic flux involves an enhanced efflux of oxaloacetate (as malate) from the mitochondria, further making acetyl CoA available for ketogenesis (as opposed to citrate production).
- 11.
Obesity is associated with the accumulation of ectopic TGs in skeletal muscle and liver. Byproducts of TG synthesis and turnover (especially diacylglycerol and ceramide) activate signaling pathways (serine/threonine kinases) that phosphorylate and desensitize the insulin receptor and insulin-receptor substrate.
Chapter 4
- 1.
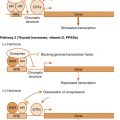
1,25-Dihydroxyvitamin D directly represses PTH gene expression. 1,25-Dihydroxyvitamin D also increases gene expression of the CaSR, which represses PTH in response to elevated serum calcium. Therefore loss of 1,25-dihydroxyvitamin D would lead to an increase in PTH secretion. PTH would also increase in response to decreases in serum Ca 2+ resulting from less efficient absorption by the GI tract in the D-deficient state.
- 2.
Vitamin D deficiency would cause increased PTH secretion by the mechanisms outlined earlier in the text. The increase in PTH secretion would maintain serum Ca 2+ levels (except in the case of rickets, where skeletal calcium stores may be limited), but would inhibit phosphate reabsorption by the proximal tubule. The resulting hypophosphatemia and decrease in the Ca 2+ × Pi product could inhibit bone mineralization resulting in osteomalacia. In rickets, the inability to mobilize Ca 2+ from bone could result in hypocalcemia as well.
- 3.
Osteoclasts perform the bone resorption phase of bone remodeling. Osteoblasts promote the differentiation of monocyte-macrophage lineage cells into osteoclast precursors (through secretion of M-CSF) and the maturation of osteoclast precursors into actively resorbing osteoclasts (through membrane expression and secretion of RANKL). Note that the PTH1R is expressed by osteoblasts, not osteoclasts.
- 4.
PTH-related peptide (PTHrP) binds to and activates the same receptor as PTH, i.e. PTH1R. PTHrP normally acts as a paracrine factor. However, high levels of PTHrP can be produced by neoplasms, thereby causing hypercalcemia (as in hyperparathyroidism).
- 5.
RANKL binds to RANK on osteoclast progenitors to promote osteoclast differentiation and also prolongs mature osteoclast lifespan and activity. OPG acts as a decoy receptor for RANKL, thereby inhibiting osteoclast-mediated bone resorption. An increase in the RANKL/OPG ratio would therefore cause excess bone resorption and reduced bone density.
- 6.
FGF23, which is produced by osteocytes, acts on the proximal tubule of the kidney to inhibit Pi reabsorption, thereby lowering serum Pi levels.
- 7.
Primary hyperparathyroidism will be characterized by elevated levels of serum PTH in the face of hypercalcemia. Typically, urinary Ca 2+ will be in the high to high-normal range. In FHH, serum Ca 2+ levels will be maintained at a higher setpoint due to reduced expression of the CaSR. PTH levels, however, should be in the normal to perhaps high-normal range. Importantly, because expression of the CaSR is also reduced in the thick ascending limb of the kidney, Ca 2+ will continue to be reabsorbed there despite the hypercalcemia, resulting in hypocalciuria.
- 8.
- a.
Vitamin D deficiency > Decreased Ca 2+ absorption > Increased PTH > Ca 2+ mobilization from bone; Increased Ca 2+ reabsorption by distal tubule; Increased Pi excretion by kidney > Hypophosphatemia > Deficient bone mineralization
- b.
Hyperparathyroidism > Increased PTH > Increased 1,25-dihydroxyvitamin D; Increased serum Ca 2+ due to PTH actions on bone and kidney, 1,25-dihydroxyvitamin D actions on intestines; Decreased serum Pi due to PTH action on kidney
- c.
Hypocalcemia > Increased PTH > Increased 1,25-dihydroxyvitamin D; Increased serum Ca 2+ due to PTH actions on bone and kidney, 1,25-dihydroxyvitamin D actions on intestines; PTH inhibition of Pi reabsorption maintains normal serum Pi
- d.
Hypophosphatemia > Decreased FGF-23 > increased 1,25-dihydroxyvitamin D > Increased Pi and Ca 2+ uptake by intestines > Decreased PTH secretion to maintain normocalcemia
- a.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


