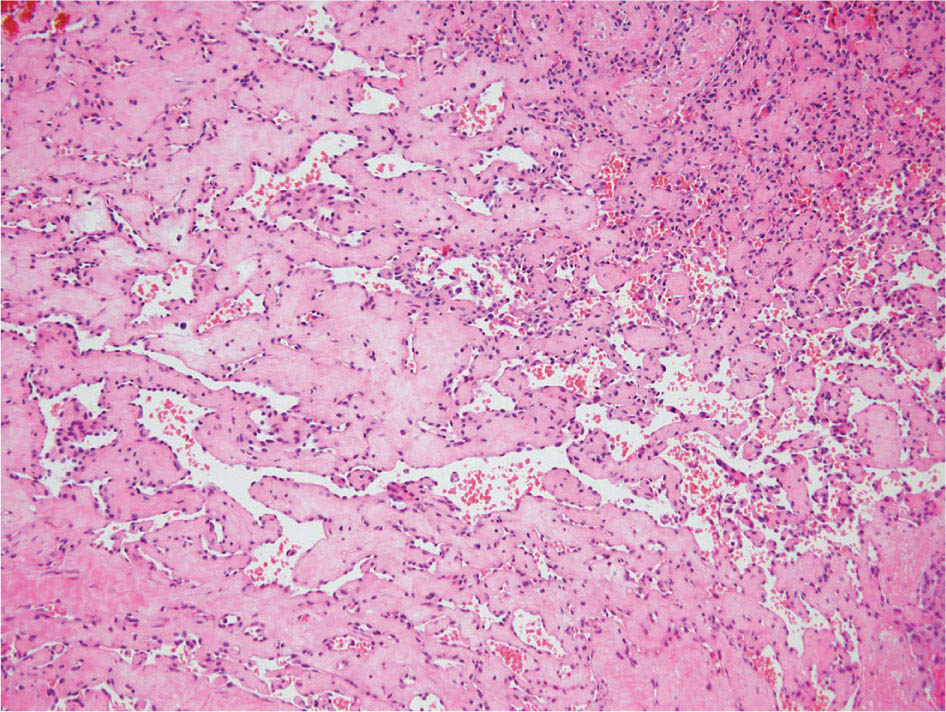
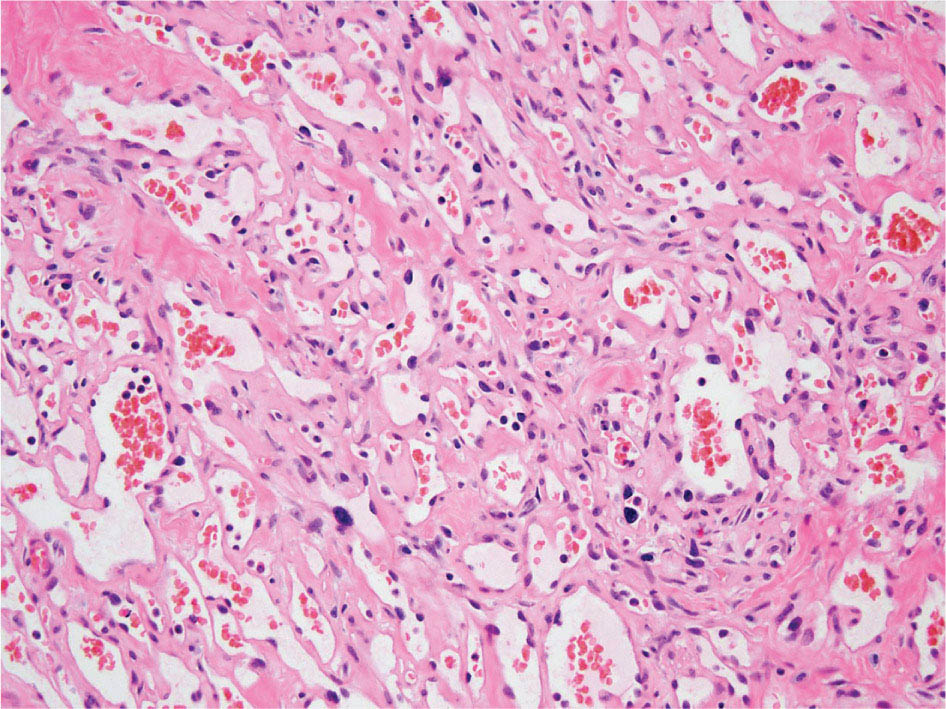
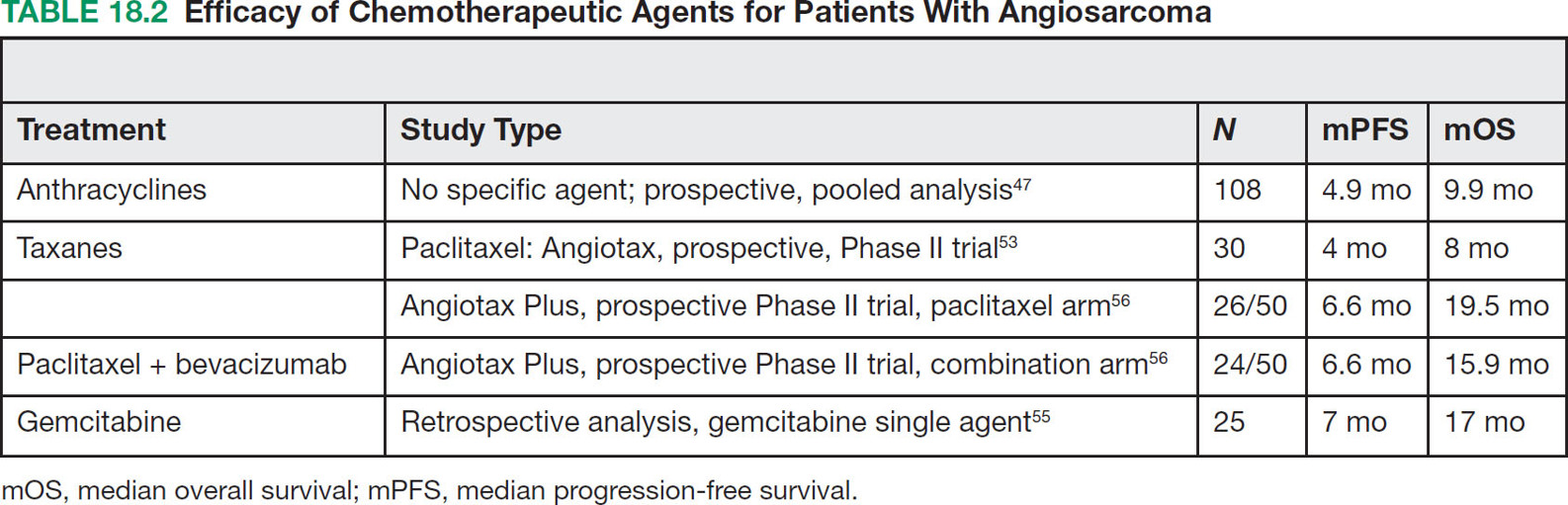
23918 Angiosarcoma Angiosarcoma (AS) is a rare tumor of endothelial origin. Despite its scarcity, AS can present with local disease in nearly any organ system. Furthermore, rate of local recurrence as well as metastasis is high, making AS a difficult cancer to treat. In locoregional disease, AS is best treated with resection as well as radiation therapy. In the setting of metastatic disease, front-line therapy with anthracyclines or taxanes has been shown to be effective. Second-line therapy with taxanes or anthracyclines has likewise demonstrated some success. Regardless of the stage at presentation, as well as treatment, long-term prognosis is poor. Significantly more research must be performed in order to better understand the biology as well as the best means of treatment for AS. This chapter reviews the clinical features, diagnostic approach, and general therapeutic approach for AS, as well as systemic therapy for metastatic AS. angiosarcoma, immunotherapy, metastasis, nonspecific β-blockers, radiation therapy, tyrosine kinase inhibitors Hemangiosarcoma, Immunotherapy, Neoplasm Metastasis, Protein-Tyrosine Kinases, Radiotherapy INTRODUCTION Angiosarcoma (AS), also known as hemangiosarcoma or lymphangiosarcoma, is a malignant tumor thought to be derived from the endothelium. The exact vascular origin is unknown, and AS likely originates from both blood vessels and lymphatics.1 AS is an incredibly rare subtype of soft tissue sarcoma (STS), accounting for <1% of all sarcomas.2 These tumors are characterized by an aggressive clinical behavior with frequent metastases and relapse.3 Given their relative scarcity, the optimal treatment of AS remains somewhat of a mystery. Furthermore, AS may present in a striking variety of different anatomic locations.4 Significant heterogeneity within the diagnosis of AS makes design and development of trials as well as therapies more difficult. As a result, there are no completed Phase III trials exclusively confined to AS, although studies are currently ongoing.5 Much of the data utilized is either Phase II, anecdotal, or part of larger studies involving STS. This ambiguity, as well as the factors already mentioned, make AS one of the more difficult cancers to treat. EPIDEMIOLOGY Collectively, ASs account for 2% of STSs.3 Given that AS is a rare subpopulation of an uncommon neoplasm, it accounts for only two to three cases per 1 million population.2 Referencing data from the Surveillance, Epidemiology, and End Results (SEER) registries, results have shown that approximately 60% of ASs are cutaneous, located within the skin or superficial soft tissues. Among primary cutaneous AS, approximately 90% occur within the Caucasian population, with a sex ratio of 1:1. Furthermore, there is a direct correlation of incidence with increased age, with most tumors occurring around age 60.6 When ASs do appear in childhood, they appear in an epidemiologic pattern different from that of adults—often with visceral as opposed to cutaneous manifestations.7 In adults, lesions are typically located on the head and neck, followed by trunk, lower extremities, and upper extremities. Despite their scarcity, AS may occur in nearly any location within the human body.4 Most AS cases occur spontaneously, but several risk factors have been identified. Risk factors for AS include radiation, chronic lymphedema, arsenic, vinyl chloride, and familial syndromes.3 Association of AS formation with lymphedema was first noted by Stewart and Treves in six women with a history of breast cancer.8 The combination of lymphedema and AS later assumed the eponymous name Stewart–Treves syndrome. Although the association of AS with prior breast cancer surgery became well recognized, AS is also associated with other forms of lymphedema.6 Numerous groups have proposed mechanisms behind the association of lymphedema and AS, including associated growth of obstructed lymphatics. Others believe that carcinogenesis occurs secondary to chronic inflammation within an immunologically privileged site that is unable to adequately remove neoplastic cells.9 Within the breast cancer population, radiation has also been shown to be associated with development of AS.10 This has increasingly been seen in women, due to radiation as part of breast-conserving treatment for breast cancer.11 The frequency of their association has bred definitions for their diagnosis. Tumors must be biopsy-proven, arising within a prior field of radiation, occurring within a latency period of several years, and arising in an area without a predominance of lymphedema.12–14 While the breast cancer survivor population is best studied, AS is also present in a large series of childhood cancer survivors.3 Interestingly, despite the association of radiation with AS, sun exposure has not been demonstrated to contribute to increased incidence.15 Other noteworthy causes of AS include toxins, such as vinyl chloride, thorium dioxide, and arsenic, which are associated with the formation of hepatic AS.16 Anabolic steroids have also been associated.4,6,17 240There have been case reports of AS arising in association with prosthetic joints18–20 as well as vascular grafts.21–23 There is insufficient data to draw meaningful conclusions about the causality of the association of AS in these cases, however. In addition to environmental exposures, several genetic abnormalities have also been linked to AS. Associations include BRCA1 and BRCA2, NF1 (neurofibromatosis type 1), IDH1 (Maffucci syndrome), and PIK3CA (Klippel–Trenaunay syndrome).3 Of note, AS is not normally associated with Li–Fraumeni syndrome, although the syndrome is associated with an increased risk of sarcoma.24 CLINICAL FEATURES AND PRESENTATION Often, cutaneous AS may resemble an ecchymotic lesion or a raised purple papule that is multifocal and may easily be mistaken for a simple benign growth. Others may present with intermittent bleeding, edema, and pain.6 These lesions typically develop on the head, neck, and face—particularly on the scalp and forehead.6,25 As these lesions grow, edema, fungation, ulceration, and hemorrhage can develop. Even early on, AS may be multifocal—which complicates therapy and results in suboptimal initial management in many cases.26 As described in larger case series of AS, often patients would present with an asymptomatic mass or lesion. Fewer had discomfort associated with the mass or lesion.4 The median size of primary tumors was 3.8 cm in one of the larger case series. Primary tumors of deep soft tissue or organs were located on extremities (10/22), trunk (3/22), heart (3/22), head/neck (2/22), liver (1/22), spleen (1/22), and adrenal gland (1/22).4 At presentation, the primary lesion is multifocal in 10% to 15% of cases. Lymph node (LN) involvement is present at initial diagnosis in 10% to 15% of soft tissue AS, in about 5% of scalp AS, and rarely in primary breast AS. About 15% of patients present with metastatic disease at the time of diagnosis.6,27 In a review of 82 patients with AS, 44% presented with locally advanced or unresectable disease.4 The 5-year overall survival (OS) rate of nonmetastatic AS is about 31% to 43%.28 OS is highly correlated with the site of the primary AS. For instance, 10-year survival rate is about 28% for extremities, 21% for scalp, and 0% for other primary sites.6 In a paper by Sinnamon et al. published in 2016, analyzing the OS of 821 patients with soft tissue and cutaneous AS undergoing resection, a more comprehensive analysis of risk factors was performed.29 Factors independently associated with worse OS included increased age, black race, head and neck location, increasing size, and positive margins. Of note, qualification of this model was performed in conjunction with the American Joint Committee on Cancer (AJCC) Cancer Staging Manual, 7th edition, which bears some differences from the 8th edition.30 DIAGNOSTIC APPROACH STSs, including AS, are staged using the AJCC system.31 Given that AS itself only represents a small proportion of STS, this becomes a relatively imperfect model.29 Clinical staging, as with any neoplasm, involves definition of the AS by physical examination, imaging, and diagnostic biopsies of related sites. It is based on characteristics of the tumor (T), nodes (N), metastases (M), and grade (G). Grading, in particular, is not recommended within AJCC Cancer Staging Manual, 8th edition. Tumors that may appear low grade may behave clinically aggressive, and many sarcoma experts approach them as they would a high-grade sarcoma. Histopathologic identification of AS is sometimes challenging because it can be mistaken for other vascular tumors. Histologic features can vary within and between cases. In a low-grade form, AS may resemble hemangioma, whereas the high-grade form may be more similar in appearance to epithelial carcinomas.3 Diagnosis of AS is typically made by resection or biopsy. Recent studies have shown that diagnosis of AS may be made by fine-needle aspiration (FNA), but cytologic features may be inadequate for proper diagnosis, so most sarcoma pathologists prefer a core-needle, incisional, or excisional biopsy.32 AS histologically has a wide variety of appearances, ranging from clearly vasoformative to poorly differentiated solid tumors. Of those that are nonvasoformative, they may appear epithelioid, as spindle cells, or mixed.33 Reticulin staining, in addition to immunohistochemistry may be useful in determining the origin of less characteristic AS. A high proportion stain for CD31, von Willebrand factor, or, albeit less specific, CD34.1 IMAGING The size of the primary lesion, and presence or absence of distant metastases need to be determined prior to treatment initiation. Diagnostic assessment, in addition to tissue assessment (Figures 18.1 and 18.2), should include MRI.3 MRI features show T1 intermediate intensity and high T2 signal 241intensity—which may be consistent with hemorrhage. The most diagnostic finding is the presence of high-flow serpentine vessels.34 AS demonstrates enhancement after intravenous contrast administration. Otherwise, imaging findings are largely nonspecific, and differential diagnosis may include glioma, cavernoma, and hemorrhagic metastases. FIGURE 18.1 Infiltrative and dissecting growth of irregularly shaped and anastomosing vascular channels are present within collagenous connective tissue—this is a characteristic feature of angiosarcoma and is viewable with hematoxylin and eosin staining (H&E, x100). FIGURE 18.2 Higher magnification of hematoxylin and eosin staining (H&E, 200x) of angiosarcoma specimen from Figure 18.1 shows clear nuclear atypia. Chest imaging, particularly CT of the thorax with contrast agent, should be performed prior to initiation of therapy to exclude metastatic disease, as the lung is the most common site of hematologic spread.4 PET/CT can be considered for patients considered at risk for LN metastases. Patients with cardiac AS are at increased risk for intracranial dissemination and MRI of the brain should be considered.35 MOLECULAR ABERRATIONS Over the last decade, our understanding of cancer genomics, not merely that of AS, has evolved significantly. We now understand that AS is a complex group of tumors with numerous associated genetic alterations. There is no one change that leads to development of all AS. In 2014, investigators found that protein tyrosine phosphatase receptor type B and phospholipase C gamma 1 are recurrently mutated in AS.36 Previous studies had shown that knockdown resistance (KDR) mutations are present in 10% 242of patients with AS.37 MYC and FLT4 mutations are seen in patients with AS secondary to radiation and lymphedema.38 A 2015 sequencing study demonstrated that over half of AS showed genetic alterations in the mitogen-activated protein kinase (MAPK) pathway, involving KRAS, HRAS, NRAS, BRAF, MAPK1, and NF1. The most frequent genetic mutation was TP53 with 35% of tumors.39 In a 2015 study of 203 adolescent and young adult sarcomas, three NTRK1 fusions were found in three tumor types, including AS.40 This is believed to be an underestimate of total prevalence and may have important implications for targeted therapy. GENERAL THERAPEUTIC APPROACH There are, as of yet, no randomized controlled trials involving the treatment of localized AS. Most treatment strategies have been developed through retrospective case series. Localized AS is initially treated with surgical resection, with the goal being R0 margins, or margins shown to be clear microscopically. Involved margins (R1 or R2) are common secondary to the multifocal nature of AS. If R1 or R2 margins are obtained, guidelines recommend repeat resection for R0 margins. Excision is often difficult, with closure requiring reconstruction rather than primary closure. In one series, the average surgical defect left after excision was 14.3 cm by 11.8 cm, with the largest spanning 28 cm by 27 cm.41 Retrospective analyses of different surgical techniques have not clearly shown superiority of any surgical approach. At a single center, there was no significant difference in OS shown between Mohs surgery and wide local excision.33 Given high risk of local recurrence, adjuvant radiotherapy is recommended, and may even be associated with a reduced risk of death (Table 18.1).4,6,41 At one center, only five of 36 patients managed initially with surgery alone were rendered disease free, versus nine of 20 who received surgery as well as radiation therapy (RT).6 In a series of 82 patients, in which 46 underwent resection of primary disease, 2/3 received adjuvant radiation. No difference in recurrence was noted between groups.4 In a retrospective analysis of 48 patients with scalp and face AS, OS rate was 45.8% for 2 years in patients who received radiation as well as surgery.42 For those patients who had surgery, or radiation alone, OS rate was 11.1% at 2 years. This population was abnormal, however, in that 45 of 48 patients had recurrence. Although there are no prospective clinical trials of adjuvant chemotherapy in AS patients, some sarcoma centers generalize from randomized clinical trials and meta-analyses that support the use of adjuvant/neoadjuvant chemotherapy for patients with high-grade sarcoma.17 This approach would add three to six cycles of doxorubicin 75 to 90 mg/m2 by 72-hour infusion plus ifosfamide 8 to 10 g/m2 divided over 4 to 5 days to resection and/or radiation. Although a large, multicenter series suggested a benefit for the addition of a taxane to radiotherapy for cutaneous AS, other limited retrospective data seem to suggest no benefit.4,43,44 Surveillance Follow-Up for Localized Disease Given the propensity of AS to recur, adequate surveillance is an important aspect of management. Although the imaging properties of AS have been studied, no definitive guidelines particular to this tumor type have been recommended.34 Follow-up imaging may be pursued in accordance with National Comprehensive Cancer Network (NCCN) guidelines for STS. For stage I disease, at least annual chest imaging should be performed, with chest CT being preferred. For stage II/III disease, reimaging after surgery, in addition to chest imaging every 3 to 6 months for at least 2 to 3 years is recommended. In one case series of 46 patients with localized disease, 26% had local recurrence after a median interval of 11 months (range of 1–63), and 43% had a distant recurrence at median of 8 months.4 In another series, in which 56 patients underwent local excision, 24 developed local recurrence alone, 18 developed local recurrence and distant metastases, and 10 developed distant metastases alone.6 243Factors associated with increased risk of recurrence included breast location, size >5 cm, and incomplete resection.45 METASTATIC DISEASE In two larger retrospective studies, between 13% and 43% patients presented with metastatic disease.4,6,33 As mentioned above, distant recurrence is not uncommon with AS and has been noted in numerous retrospective studies. Sites of distant metastases include the lung, bone, bowel/mesentery, liver, regional LNs, skin, brain, adrenal glands, kidney, and heart.33,45 In one study of 99 patients with metastatic AS, diseas-specific survival rates were 40%, 29%, and 10% for 1, 2, and 5 years, respectively.45 Overall, the outcomes for patients with metastatic AS are significantly worse than those for patients with local tumors or locally recurrent tumors. SYSTEMIC THERAPY FOR METASTATIC DISEASE Anthracyclines In general, metastatic AS is considered to be a fatal and incurable disease. While there is no true standard of care, studies have been able to demonstrate the effectiveness of numerous cytotoxic and targeted therapeutic agents. Current regimens largely involve anthracyclines or taxanes.46 In 2014, a pooled analysis of AS patients from the European Organization for Research and Treatment of Cancer (EORTC) clinical trials of first-line anthracycline-containing regimens showed similar outcomes when comparing AS to other STS histologis types.47 A total of 108 patients with AS were included in the analysis, with 25% response rate achieved compared to 21% of the remaining STS patients. Progression-free survival (PFS) and OS were similar for AS and other STS histologic types. Median PFS for AS patients was 4.9 months, with a median OS (mOS) of 9.9 months. This is comparable to a median PFS (mPFS) of 4.3 months and OS of 12 months for other STSs. Only 41% of AS patients survive to 1 year, compared to 50% of other STS patients. In 2016, olaratumab, in combination with doxorubicin, was approved for treatment of STS.48 Olaratumab is a monoclonal antibody against platelet-derived growth factor receptor A (PDGFRα). A Phase II study, incorporating unresectable and metastatic STSs, randomized 1:1 to doxorubicin/olaratumab and doxorubicin alone, showed significantly increased median overall survival (mOS) with addition of olaratumab. Of note, the study population contained a total of seven AS patients and was consistent across subtype stratification (leiomyosarcoma vs. nonleiomyosarcoma). OS was only significantly improved in the first-line setting—meaning that the combination of olaratumab and doxorubicin should be utilized in the first-line setting. Unfortunately, the Phase III study did not confirm the efficacy of olaratumab, and this agent is no longer available. In a retrospective study at one center, approximately 21 patients received pegylated liposomal doxorubicin (PLD) in the first-line setting.49 Within this study, the response rate was similar between PLD, doxorubicin, and taxane agents within the first-line setting. Given this data, PLD may be a reasonable treatment strategy for a patient with AS, in order to mitigate the toxicity of doxorubicin. Taxanes Historically, sensitivity of AS to taxanes has distinguished them from other STS subtypes.50 Numerous studies have demonstrated the effectiveness of taxanes in the first-line setting.51,52 In particular, the Phase II ANGIOTAX trial showed an overall response rate (ORR) of 19%, with mPFS of 4 months, and an OS of 8 months with weekly paclitaxel.53 Results from the ANGIOTAX trial were in line with prior retrospective analyses, showing similar PFS, but variable ORR. Gemcitabine Gemcitabine has been shown to be effective when combined with docetaxel as a second-line regimen for most STSs.54 A retrospective study of 25 patients with advanced AS demonstrated an ORR of 68% and a mOS of 17 months.55 While mostly studied in combination with anthracyclines or taxanes, gemcitabine may also be considered as single-agent treatment (see Table 18.2).55,56 In a retrospective analysis of 25 patients who received weekly gemcitabine alone, patients had an ORR (partial response + complete response) of 68%. mOS in this case series was 17 months with mPFS of 7 months. In one patient, a locally advanced 244tumor was made resectable again.55 These results were highly encouraging, particularly in light of the tolerability of the study agent.

Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


