Angiogenesis
11.1 INTRODUCTION: THE TUMOR–VASCULAR INTERFACE
An important feature of malignancies is the associated emergence of new and abnormal contact points between cancer cells and the various facets of the host vascular system (Folk-man and Kalluri, 2003; Kerbel, 2008). Prior to transformation many normal epithelial tissues (eg, in the gut, skin, and exocrine glands) are functionally linked to, but often anatomically separated from, the vasculature, for instance, by basement membranes and connective tissue layers (Rak, 2009). These barriers are compromised during the course of the malignant process, resulting in abnormal, often direct and reciprocal interactions between vascular components (endothelial cells, blood cells, plasma, or lymph) and cancer cells at this new tumor–vascular interface.
Tumor–vascular interactions are important for disease progression, because of several “outside-in” effects, such as supply of oxygen, nutrients, growth factors, metabolites, paracrine and adhesive tumor–vascular interactions, recruitment/retention of the host immune, inflammatory, and bone marrow-derived progenitor cells, as well as delivery of drugs, hormones, and regulatory molecules. The vascular interface also mediates important “inside-out” processes, notably, intravasation of metastatic cancer cells, emission of angiogenesis-regulating, proinflammatory, procoagulant, hormonal, and metabolic (eg, cachexia-inducing) signals, as well as shedding of tumor-related microvesicles (exosomes) containing biologically active molecules (Rak, 2009). The nature of the tumor–vascular interface is influenced by a succession of genetic and epigenetic alterations in cancer cells, microenvironmental influences (hypoxia, inflammation), as well as host genetic background, accompanying diseases (comorbidities), ageing, and other factors (Kerbel, 2008).
The term angiogenesis and its evolving meaning have a long history. The term was first used by John Hunter in 1787, and later reintroduced in 1935 by Artur Tremain Hertig, to describe non-cancer-related blood vessel growth processes (Roy-Chowdhury and Brown, 2007). Works of Lewis (1927), Sandison (1928), Ide (1939), and Algire (1945), as well as Greenblatt and Shubik (1968) gradually led to a description of vascular expansion associated with a developing cancer, along with some of the first experimental approaches to study the related processes. It was not until the early 1970s that the concept of targeting angiogenesis for therapeutic purposes (anti-angiogenesis) was proposed by Judah Folkman (Folkman, 1971). This form of therapy is now a part of the clinical management in several malignancies. Further development of these approaches depends on improving our understanding of mechanisms governing the response of the vascular system to an emerging malignancy, and the relationships, both local and systemic, between cancer cells and the vascular and non-vascular host tissues (Folkman, 2007).
11.2 CONSTITUENTS OF THE VASCULAR SYSTEM
11.2.1 Blood Vessel and Lymphatic Networks
Blood circulation ensures the delivery of oxygen, nutrients, macromolecules, hormones, and cells (eg, immune, inflammatory, or stem cells), to the vicinity of every living cell, while removing catabolites and waste products. To remain viable, each mammalian cell must be located no further than 100 to 180 μm from the nearest functional (perfused) blood vessel capillary. Directionality, efficiency, and organ-specificity of the blood flow are developmentally preprogrammed by the geometry, physical properties, and hierarchical architecture (arborization) of the vascular system. Thus, cardiac output is directed through the tree of arteries of decreasing caliber and changing wall structure, including elastic, muscular-type, and contractile arteries, as well as smaller and precapillary arterioles, which eventually branch into capillary blood vessels. Capillaries converge to form the postcapillary venules, followed by small, midsize, and large veins that differ from their corresponding arteries by lower pressure, velocity, and content of blood (eg, low oxygen levels), as well as by thinner wall structure and (in some segments) the presence of intraluminal valves to maintain flow directionality.
Each blood vessel is composed of a crucial inner lining (endothelium) made of endothelial cells, which are surrounded by 1 or more supportive layers containing cells with contractile characteristics of smooth muscle (mural) cells, sheaths (laminae) of extracellular matrix (ECM) and other components (eg, innervation). Collectively, these structures (Fig. 11–1) provide an inner surface that is resistant to coagulation, serve as a source of intercellular and biomechanical signals required for endothelial cell survival, and afford physical support and contractility of mural cells, as appropriate for the vessel caliber and site-dependent function (Carmeliet and Jain, 2011).
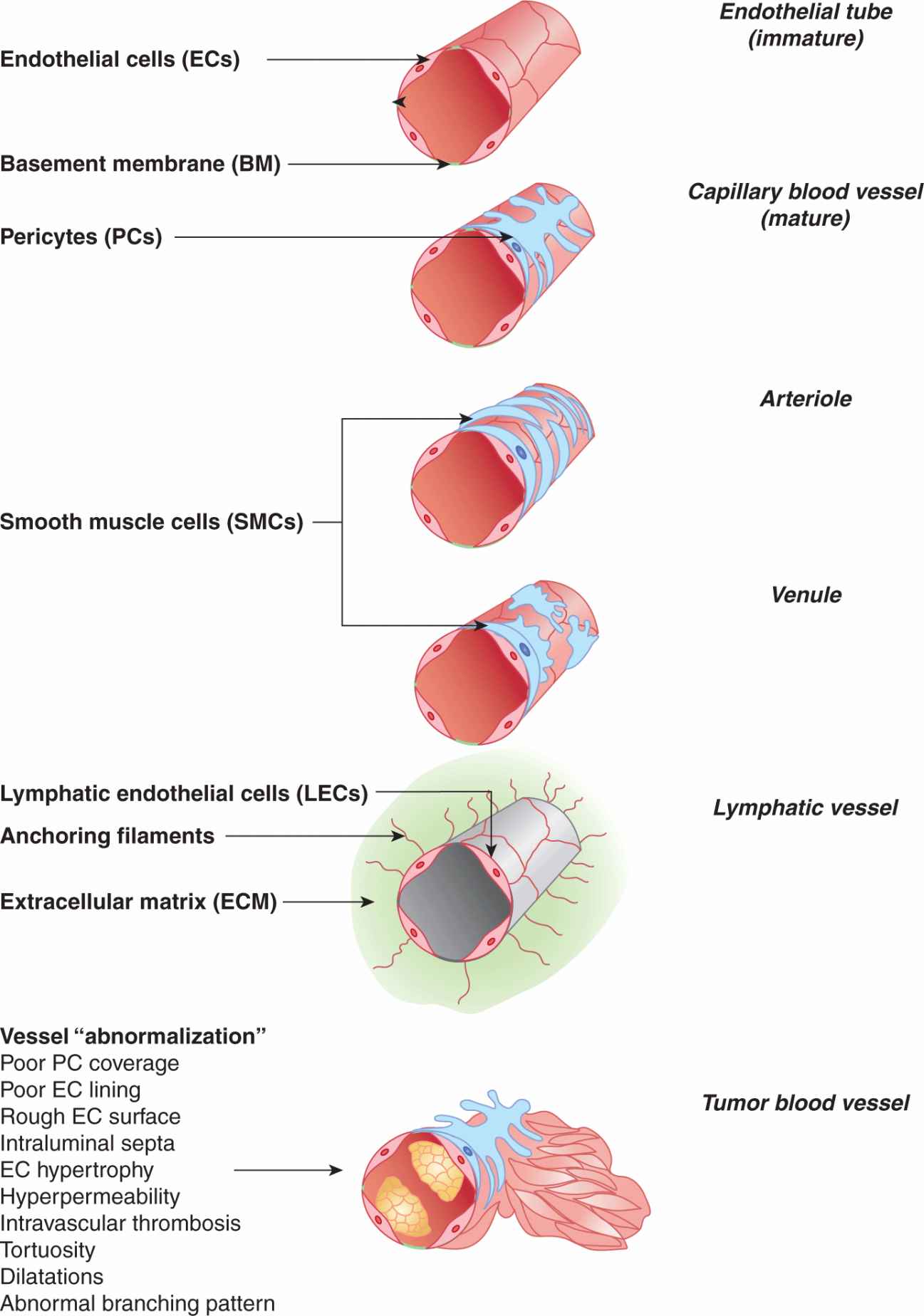
FIGURE 11–1 Structural constituents of the microvasculature. Cellular architecture of the main types of microvessels, including a newly formed endothelial tube (top), capillary blood vessels, precapillary arteriole, postcapillary venule and lymphatic capillary. Blood vessel endothelial cells (ECs), pericytes/smooth muscle cells (PCs/SMCs), and lymphatic endothelial cells (LECs) are the key constituents of the microvasculature, including in cancer. Tumor blood vessels exhibit multiple structural and functional abnormalities, and features consistent with lack of maturation/normalization, hence referred to as vessel abnormalization (see text for details).
Capillary blood vessels (Fig. 11–1) constitute the main point of molecular exchange between tissues and the circulating blood. Capillaries are thin walled tubes of blood endothelial cells (ECs), with lumen 8 to 20 μm in diameter, and exterior wrapped in collagen type IV-rich basement membranes, which also envelope discontinuous layers of smooth muscle-like cells (SMCs), which at the capillary level are known as pericytes (PCs). The permeability of EC layers for molecules is tightly regulated. Thus, while capillaries in most of the vascular beds are permeable to fluids and certain micromolecules (oxygen, certain ions), but highly restrictive to macromolecules, in the brain such passage is controlled even more tightly, a property known as the blood–brain barrier (BBB), while being less restrictive in endocrine organs (Carmeliet and Jain, 2011).
Fluid released (extravasated) through the capillary walls into the intercellular tissue space (interstitium) is collected by lymphatic capillaries (lymphatics) (Tammela and Alitalo, 2010). Lymphatic vascular networks are present in almost all organs, except for the central nervous system (CNS), bone marrow (BM), and avascular tissues, such as cartilage, cornea and epidermis. Lymphatic endothelial cells (LECs) are connected to the surrounding ECM by specialized fibrillin-containing anchoring filaments (Alitalo et al, 2005). Capillary lymphatics are open ended and thin walled, have discontinuous or absent basement membrane, and lack PCs, all of which predisposes them to efficiently “collect” interstitial fluid, macromolecules, and cells (see Fig. 11–1). Lymphatics drain their content (lymph) to the regional lymph nodes, while the larger lymphatic channels subsequently converge, forming a common central duct (thoracic duct), which carries the lymph to the left subclavian vein (Tammela and Alitalo, 2010). Thus, in adults 1 to 2 L of interstitial fluid containing molecules and cells enter the lymphatic system per day, and eventually reach the general circulation. In this manner also, metastatic cancer cells may reach lymph nodes and the blood circulation (Tammela and Alitalo, 2010).
11.2.2 Properties of Cells Involved in Vascular Structures
Several populations of cells contribute to the maintenance, growth, and remodeling of vascular structures (Fig. 11–2); they include resident endothelial (ECs, LECs) and mural cells. The latter include SMCs in larger vessels, and PCs in capillaries, permanently incorporated into the vascular wall. Bone marrow-derived cells (BMDCs) may also take transient residence within (around) the vascular wall, as is the case for several subsets of progenitor and regulatory cells of myeloid and endothelial origin (De Palma and Naldini, 2006).
Endothelial cells line the entire inner surface of the circulatory system. In this fashion, they control the properties of blood and those of perivascular tissues. In the adult, endothelial cells are usually quiescent (more than 99.9%), with a cell turnover time ranging between 6 weeks and many years, and somewhat faster at vascular branching points; exceptions include wound healing and cyclical changes in female reproductive organs. Endothelial cells exhibit several common properties, such as flat morphology, anticoagulant luminal surfaces, and the expression of certain specific molecules (pan-endothelial markers), which include CD31 (PECAM), CD144 (VE-cadherin), von Willebrand factor (vWF), CD202b (TIE2), CD34 (sialomucin), CD146 (P1H12 antigen/MUC18), and CD105 (endoglin). However, endothelial cells may also display regional differences dependent on vessel type, caliber, and organ site (Folkman and Kalluri, 2003).
Mural cells enforce the integrity of the endothelial tube. These elongated and contractile cells provide blood vessels with mechanical resistance, functional stability and vasoconstrictive properties (Carmeliet and Jain, 2011). In capillaries, PCs form processes that extend along the vessel axis making contacts with several endothelial cells. In this manner endothelial cells receive survival signals and become less dependent on soluble mediators. Mural cell-specific antigens (markers) include α smooth muscle actin (αSMA), desmin, CD13, 3G5 ganglioside, CD248 (endosialin), NG2 chondroitin sulphate proteoglycan, RGS5, and platelet-derived growth factor receptor beta (PDGFRβ). PCs are actively recruited to, and retained at, the developing vessel wall through signaling mechanisms involving angiopoietin 1 (ANG1) and its endothelial receptor (TIE2/TEK), as well as through action of multiple other factors (Yancopoulos et al, 2000; Jones et al, 2001).
Bone marrow-derived regulatory and progenitor cells maintain vascular homeostasis at the systemic level (see Fig. 11–2). Local cytokine release leads to the recruitment and retention of various populations of BMDCs. These include endothelial progenitor cells (EPCs), also known as circulating endothelial progenitors (CEPs), to distinguish them from more differentiated circulating endothelial cells (CECs) that may slough off the vessel wall as a result of damage, or cell death (Bertolini et al, 2010). EPCs/CEPs have been implicated in formation of new blood vessels, vascular regrowth, and metastasis, but their ability to differentiate into mature ECs and contribute to the vessel wall still remains controversial. They may, however, play a role in maintenance of the existing endothelial layers (Xu, 2006). Tumor-associated endothelial cells were recently found to contain a subset of cells with multipotential (stem-like) properties and the capability to differentiate into chondrocytes and bone cells, and to trigger calcification (Dudley et al, 2008).
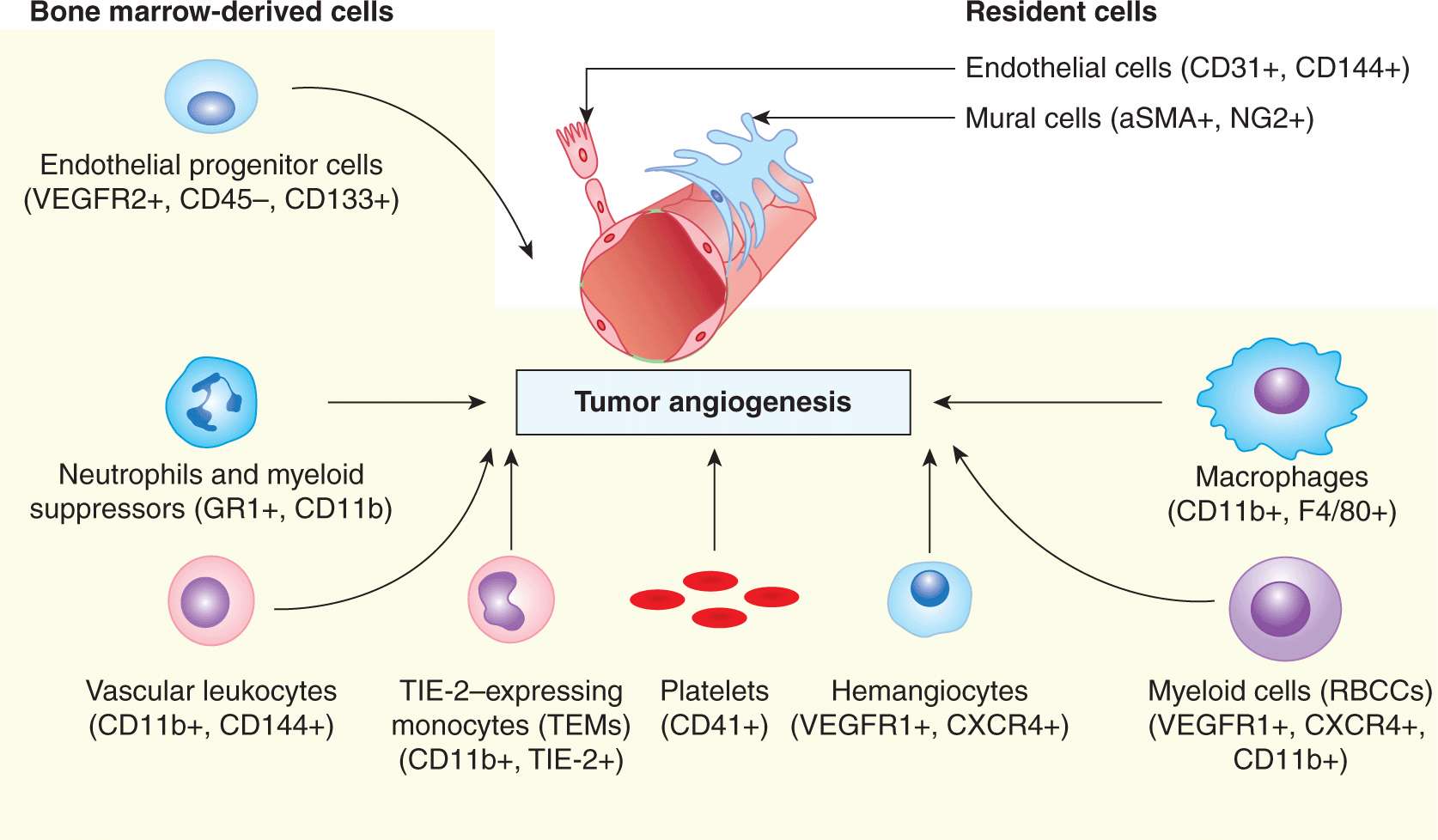
FIGURE 11–2 Cell populations involved in tumor angiogenesis. Vascular growth is coordinated by structural resident cells in the preexisting blood vessels, as well as by several populations of circulating cells (shaded area), mostly of bone marrow origin. Although the structural contribution of endothelial progenitor cells (EPCs) in tumor neovascularization remains controversial, the regulatory function of these cells and that of several types of myeloid cells is increasingly well established, for example, as sources of vascular endothelial growth factor (VEGF), alternative angiogenic factors, guidance signals, proteolytic activity, and other effects. Angiogenic fibroblasts, cancer cells, and immune effectors are not included in this diagram for simplicity (see text for details).
In addition to EPCs, several types of myeloid cells play regulatory roles during remodeling and maintenance of the vasculature, including changes occurring in the course of cancer. The best-described subpopulations of these cells encompass: (a) tumor-associated M2-type macrophages, characterized by the expression of F4/80 and CD11b antigens (F4/80+/CD11b+); (b) monocytes expressing TIE2 receptor (TIE2+/CD11b+); (c) recruited bone marrow circulating cells (VEGFR1+/CXCR4+/CD11b+); (d) hemangiocytes (VEGFR1+/CXCR4+); (e) vascular leukocytes (CD11b+/VE-cadherin+); and (f) angiogenic neutrophils (GR1+/CD11b+). Proangiogenic properties have also been ascribed to other myeloid cells, such as mast cells (MCs), dendritic cells (DCs), and hemopoietic stem cells (HSCS) (De Palma and Naldini, 2006; Ahn and Brown, 2009). Several of these cell types not only produce angiogenic factors (eg, vascular endothelial growth factor [VEGF]-A or Bv8 [Bombina variegata-secreted protein 8]), but also express overlapping markers with endothelial cells (Bertolini et al, 2010).
11.3 PROCESSES LEADING TO BLOOD VESSEL FORMATION
11.3.1 Vasculogenesis
Vasculature is the first organ system to develop during embryogenesis, around midgestation in mice (Ema and Rossant, 2003). The process (Figs. 11–3 and 11–4) consists of at least 4 distinct phases (Carmeliet and Jain, 2011), including (a) the emergence of endothelial progenitors (angioblasts); (b) coalescence and differentiation of angioblasts to form the primitive network of endothelial tubes (vasculogenesis), a structure known as the primary capillary plexus; (c) branching of new vascular projections (sprouts) from the preformed endothelial tubes (angiogenesis); and (d) remodeling, expansion, and diversification of the vascular network to form the arterial and the venous side of the circulation, a process which defines the directionality of the blood flow.
As the vascular system matures, the ingress of mural cells (vascular maturation) stabilizes the endothelial channels. Some of these channels may be superfluous, or nonfunctional, which leads to their regulated regression (pruning). At the same time, branching and angiogenesis lead to the establishment of a vascular hierarchy where some vessels become supply lines for smaller capillaries. To meet the volumetric and mechanical requirements, these supplying vessels undergo circumferential expansion and wall remodeling, a process termed arteriogenesis (Carmeliet and Jain, 2011). Remodeling and diversification of various segments of the vascular tree serves to meet the unique metabolic, structural and mechanical requirements of different organs and tissues. Thus, the vascular architecture of the heart muscle differs from that of the liver or kidney, as well as between other organ sites (Folkman and Kalluri, 2003). Conversely, tissue growth and geometry may be influenced by the vasculature, which, for example, controls mechanisms of liver regeneration and influences organ size (Folkman and Kalluri, 2003).
11.3.2 Endothelial Repair
EPCs can be recruited to sites of pathological losses in endothelial lining (denudation). EPC-dependent endothelial repair is thought to contribute to attenuation of the vascular damage in the course of inflammatory diseases such as atherosclerosis (Xu, 2006). These cells may also home to intravascular clots and facilitate formation of inner vascular channels within them (recanalization).
11.3.3 Angiogenesis
Angiogenesis is a process whereby new vascular structures emerge from ones that have already been established in the surrounding tissues or within the tumor mass (see Fig. 11–3). In cancer, this process is viewed as the key source of vascular growth associated with tumor formation and metastasis, and one that occurs via at last 3 different mechanisms: (a) intussusception; (b) vascular splitting; and (c) sprouting angiogenesis (Carmeliet and Jain, 2011). Intussusception leads to division of a larger or dilated capillary vessel into smaller channels, as a result of external pressure exerted by extravascular tissue. Similarly, separation of the vascular lumen into several branches can be achieved by formation of intraluminal septa (splitting). Both of these processes lead to formation of additional capillary loops that could enlarge and make contacts with a greater volume of the adjacent tissue (Carmeliet and Jain, 2011).
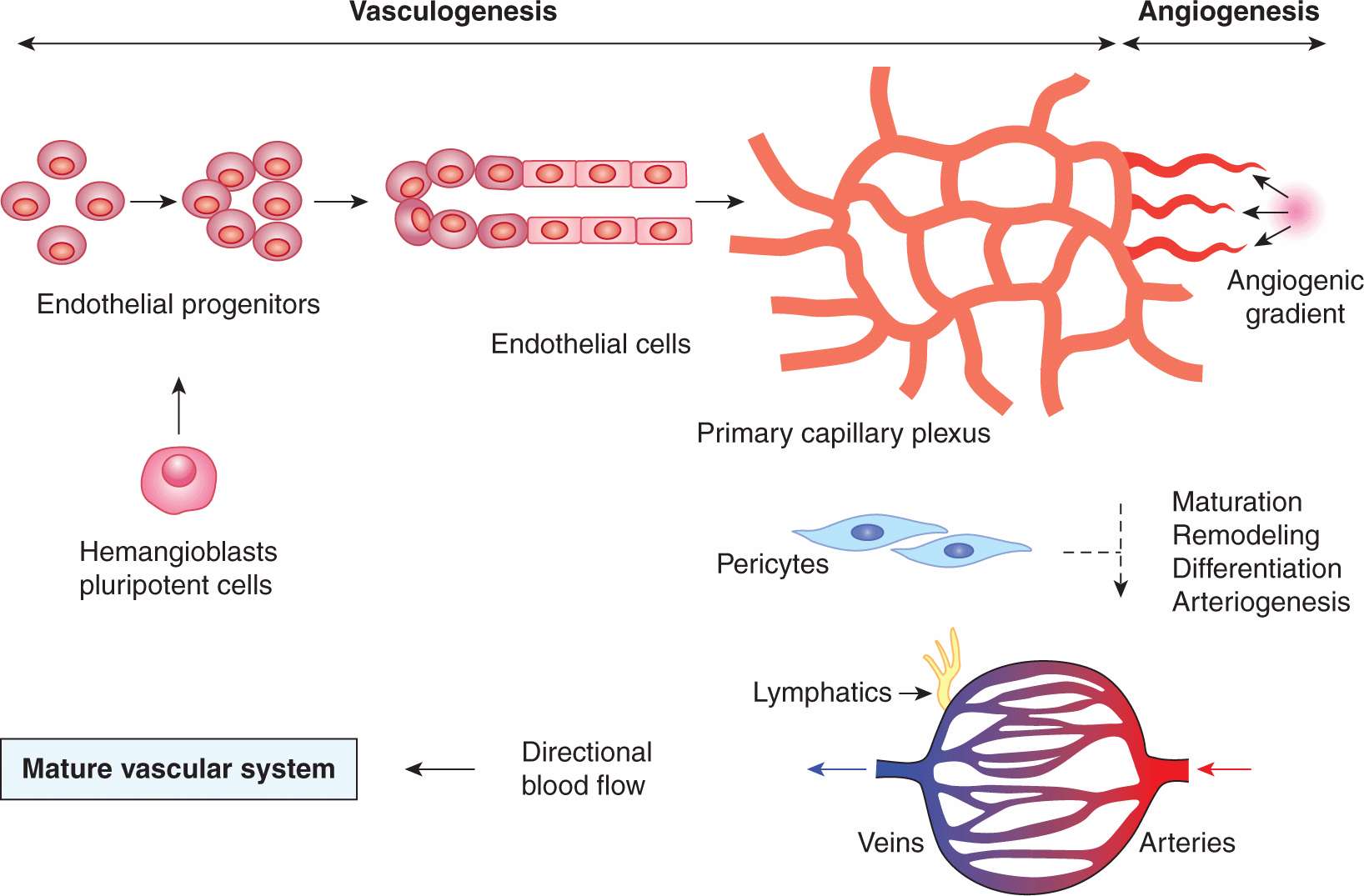
FIGURE 11–3 Development of the vascular system. A spectrum of blood vessel-forming processes is activated during development, and partially reactivated during postnatal vascular growth (wound healing, pregnancy, vascular diseases, and cancer). Endothelial cells, the key organizing element of the vascular system emerge from their precursors, which are a product of the lineage commitment of earlier precursors (hemangioblasts) in the embryonic mesenchyme, or may potentially arise through transdifferentiation of other cells. Vasculogenesis entails coalescence of endothelial progenitors, their differentiation to endothelial cells and formation of primitive vascular tubes, which organize themselves into a homogenous, directionless, and largely nonperfused network (primary capillary plexus). Angiogenesis is a process of outgrowth of vascular structures from the preexisting primary plexus, or mature vasculature. During development, angiogenesis is accompanied by the antithetical process leading to removal of superfluous vessels (pruning), both of which are essential for the establishment of the proper hierarchy (arborization) of the vascular network and for directional blood flow from arterial to the venous side of the circulation. The integrity of the emerging capillary blood vessels is ensured by arrival of PCs (vessel maturation). The arterial vessels that supply blood to the expanding microcirculation undergo a remodeling and circumferential growth processes, often referred to as arteriogenesis; see the text and references for further details. (Carmeliet and Jain, 2011; Ema and Rossant, 2003.)
Sprouting angiogenesis constitutes the main mechanism leading to the expansion of cancer-related microvascular networks (see Fig. 11–4). The exposure of a precapillary vessel to the gradient of proangiogenic activity (eg, VEGF expression induced by hypoxia [see Sec. 11.5.3 and Chap. 12, Sec. 12.2]) leads to several orchestrated responses, which begin with vessel dilatation giving rise to formation of a thin-walled, regionally distended structure known as a “mother vessel” (Pettersson et al, 2000). This transition reflects the activation state of the still intact monolayers of endothelial cells, here referred to as phalanx cells (Carmeliet and Jain, 2011). Formation of mother vessels is often followed by responses of mural cells (PCs), leading to their focal detachment from the endothelial tube (PC dropout), which thereby becomes liberated from the structural constraints, more exposed to extravascular stimuli, and capable of deployment of endothelial cells (Carmeliet and Jain, 2011). The centerpiece of these complex endothelial-PC interactions is the upregulation of angiopoietin 2 (ANG2) in endothelial cells, exposed to high levels of VEGF. In endothelial cells, ANG2 acts as an autocrine antagonist of the TIE2 receptor tyrosine kinase expressed on the surface of these cells (Fig. 11–5). In this context, ANG2 binding relieves phosphorylation of TIE2, as a result of displacement of its natural PC-derived agonist, angiopoietin 1 (ANG1). Inactivation of the ANG1/TIE2 interaction disrupts the pathway that normally mediates the stable PC-endothelial contact, and as a consequence liberates endothelial cells that may now form new outgrowths (Yancopoulos et al, 2000). An important step required for endothelial liberation is the focal dissolution of the capillary basement membrane by matrix metalloproteinases (MMPs; Fig. 11–6), which also release additional angiogenic factors from the ECM stores (Carmeliet and Jain, 2011).
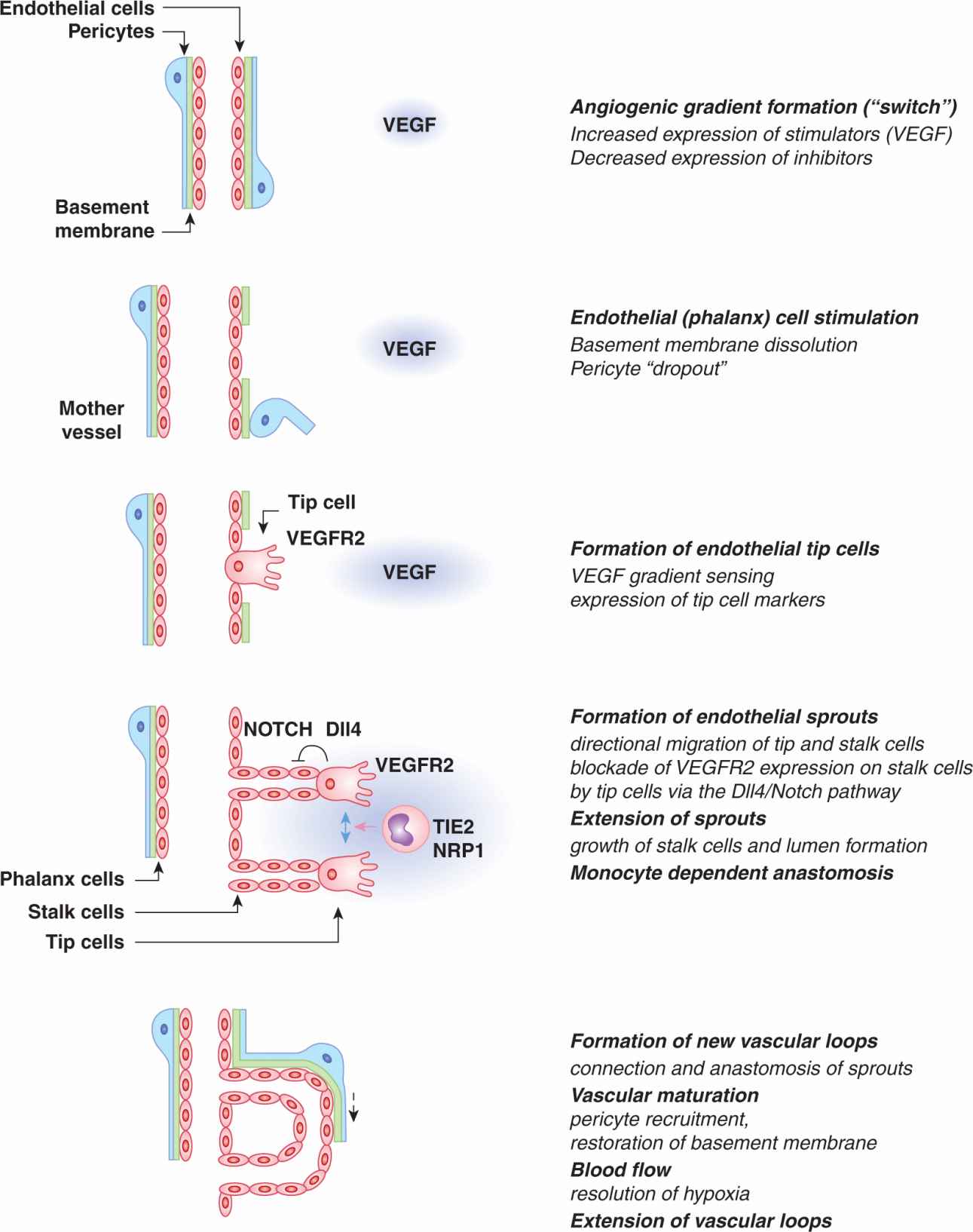
FIGURE 11–4 Sprouting angiogenesis. The change in balance between angiogenesis stimulators and inhibitors (angiogenic switch) and especially local upregulation of VEGF and formation of the VEGF gradient leads to stimulation of vascular responses known as sprouting angiogenesis. Stimulated blood vessels undergo a series of structural changes that consist of localized dissociation of pericytes (“dropout”) from the endothelial tube, dissolution of the basement membrane and recruitment of the angiogenesis-directing cells (tip cells) from the endothelial monolayer (phalanx cells). Capillaries enlarge (to form mother vessels) and deploy cohorts of endothelial cells (stalk cells) led by VEGF gradient-seeking tip cells expressing high levels of VEGFR2 and DLL4. Interaction of tip cell-related DLL4 with NOTCH on following stalk cells suppresses VEGFR2 expression on the latter cell subset and ensures that they don’t become superfluous tip cells. Vascular sprouts extend and undergo lumen formation. TIE2–expressing monocytic cells orchestrate the encounter and connection (anastomosis) of nearby sprouts to complete the formation of a functional vascular loop. Capillary loops can extend further to promote an increased tissue perfusion, a process known as looping angiogenesis, and operative during granulation tissue formation.
The exposure of endothelial phalanx cells to extravascular angiogenic gradients triggers the process of coordinated formation, movement, and extension of angiogenic sprouts (see Fig. 11–4). Each sprout is composed of a single specialized tip cell equipped with hair-like, ligand-sensing projections (filopodia), containing high concentrations of VEGF receptors (especially VEGFR2) (Gerhardt et al, 2003). Tip cells also express other molecules (eg, platelet-derived growth factor B [PDGF-B]), as well as high levels of a transmembrane protein known as Delta-like ligand 4 (DLL4), the expression of which is induced by VEGF (Noguera-Troise et al, 2006). DLL4 acts as the key ligand for the NOTCH receptor present on endothelial stalk cells that follow each migrating tip cell (see Chap. 8, Sec. 8.4.2). This interaction involving the activation of the Notch pathway suppresses VEGFR2 expression in stalk cells adjacent to a tip cell, which prevents them from producing DLL4, and thereby becoming tip cells themselves. In this manner, the Notch pathway enforces the unique identity (“leadership”) of the tip cell within the cellular hierarchy of each vascular sprout (Noguera-Troise et al, 2006). Indeed, when the DLL4/NOTCH interaction is inhibited, multiple tip cells and sprouts emerge, leading to hyperdense, nonperfused and dysfunctional capillary networks, a phenomenon known as nonproductive angiogenesis (Noguera-Troise et al, 2006). The vascular branching through formation of new sprouts may be fine-tuned with the contribution of another NOTCH ligand, Jagged 1. Jagged 1 blocks the effects of DLL4 on stalk cells leading to a controlled stimulation of processes leading to formation of additional tip cells and sprouts (Carmeliet and Jain, 2011).
Tip cells serve as guidance devices for endothelial sprouts, the numbers and directions of which they control, as they move along the path of angiogenic (VEGF) gradients (Gerhardt et al, 2003). Cohorts of stalk cells follow each tip cell and contribute to the sprout extension by directional collective migration and cell division, the latter occurring mainly at the sprout base (Gerhardt et al, 2003). Eventually neighboring sprouts connect (anastomose), a process regulated by a subset of tissue macrophages expressing TIE2 and Neuropilin 1 (NRP1) receptors (Fantin et al, 2010). Formation of endothelial sprouts is accompanied by generation of the vascular lumen. This may occur either through the merger of intracellular vacuoles within endothelial cells, or by formation of intercellular spaces between them, as they form cohorts of cells aligned in parallel (Carmeliet and Jain, 2011). Collectively, these processes of growth, lumen generation, and sprout anastomosis result in formation and extension of functional endothelial capillary loops, which may take up their roles in perfusion of new regions of the tissue or tumor mass (see Fig. 11–4).
11.3.4 Vascular Maturation
Maturation of vessels involves assembly of the mural cell layer around the newly formed endothelial tube (Carmeliet and Jain, 2011). Tumor-associated capillaries were traditionally thought to be devoid of PCs, but more recent studies confirmed the presence of these cells and their impact on the properties of the tumor microcirculation (McDonald and Choyke, 2003). Indeed, angiogenic endothelial cells, including tip cells, secrete PDGF-B, which is deposited onto the heparin sulphate proteoglycan chains and serves as a chemoattractant for regional PCs expressing PDGFRβ (Gaengel et al, 2009). PCs also secrete ANG1, which orchestrates their interactions with endothelial cells and acts as an endothelial survival factor by activating the TIE2 receptor (Hanahan, 1997; Jones et al, 2001). Vascular maturation is also critically dependent on sphingolipid signaling, notably generation of the Sphingosine 1 phosphate (S1P), which regulates N-cadherin, an adhesion molecule that links endothelial cells and PCs (Gaengel et al, 2009). Upon their attachment to the endothelial tube (see Fig. 11–1), PCs differentiate and assume a more mature phenotype, a process that is thought to be regulated by transforming growth factor beta 1 (TGFβ1) (Gaengel et al, 2009). PC coverage of endothelial structures and the vascular maturation (stabilization) processes are profoundly affected by endothelial oxygen sensor Prolyl hydroxylase 2 (PHD2) (Carmeliet and Jain, 2011), and a number of other mechanisms responsible for cellular integrity, hypoxia response, and survival (see Chap. 12, Sec. 12.2). Overall, vascular maturation provides newly formed capillaries with structural support and mechanical resistance, and reduces endothelial cell demand for soluble survival factors, such as VEGF (Carmeliet and Jain, 2011).
11.3.5 Lymphangiogenesis
Formation of new lymphatics from preexisting lymphatic vessels is called lymphangiogenesis (Tammela and Alitalo, 2010). There are only small differences in the spectrum of proteins synthesized in lymphatic endothelial cells (LECs) and vascular ECs. However, the properties of these cells, their function, and their responses to stimuli are markedly different (Alitalo et al, 2005). Studies of LECs and lymphangiogenesis have been advanced by a recent description of their distinct markers, such as the Prospero homeobox transcription factor (PROX-1), Podoplanin, and Lymphatic vessel hyaluronan receptor-1 (LYVE-1) (Alitalo et al, 2005). Lymphatics are also dependent on the activity of the forkhead transcription factor FOX2c. However, the centerpiece of the unique pathway governing lymphangiogenesis consists of two VEGF-related growth factors, VEGF-C and VEGF-D. These factors interact with a distinct receptor expressed preferentially on LECs (and on ECs in certain tumors), known as VEGFR-3/FLT-4 (Alitalo et al, 2005; see Fig. 11–5).
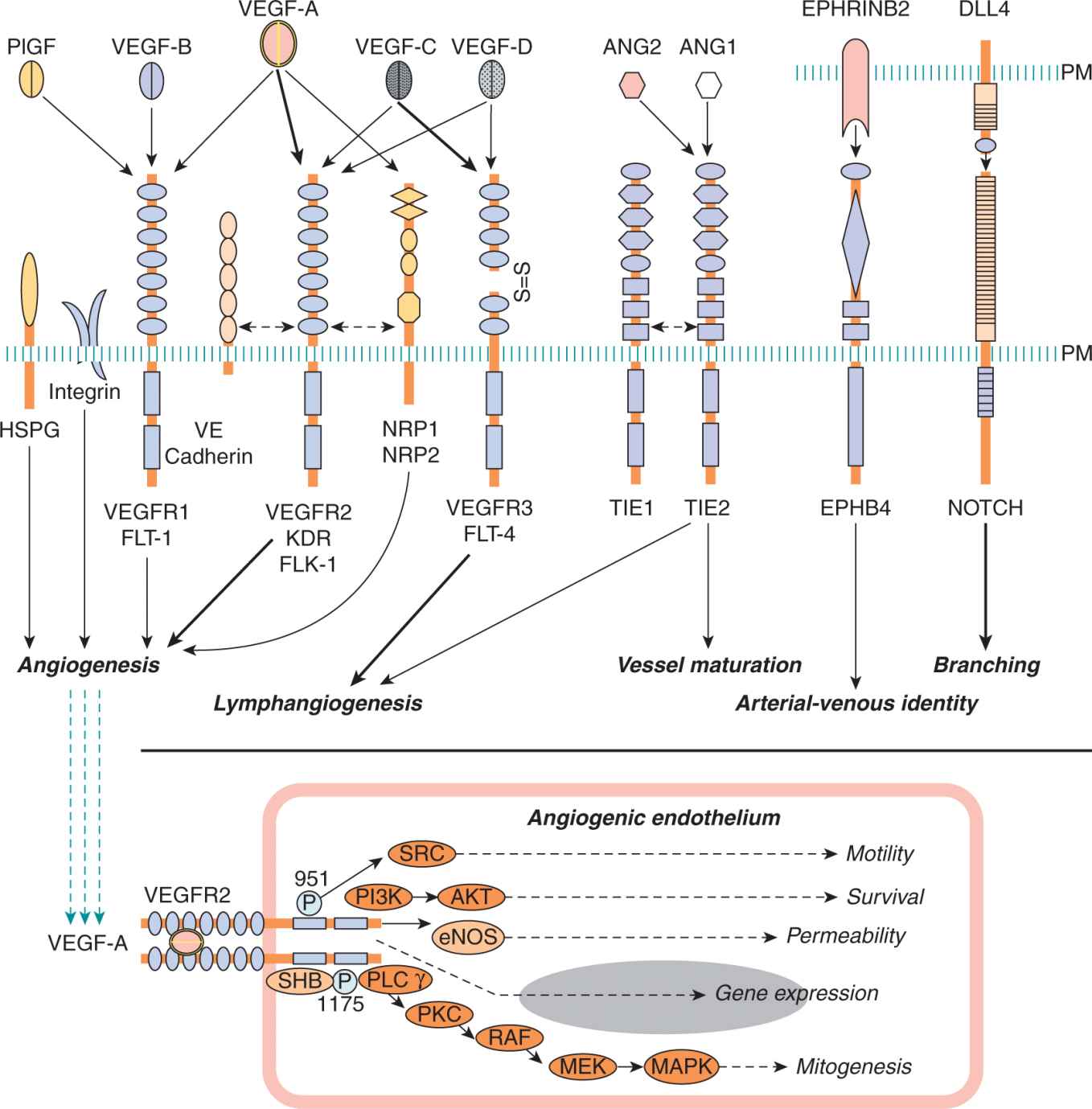
FIGURE 11–5 Some of the key elements of the signaling circuitry involved in blood vessel formation and tumor angiogenesis. Top panel: Ligands, receptors and co-receptors involved in angiogenic signaling. Note that EphrinB2 and DLL4 are transmembrane ligands. Domain structure, signaling properties, targets, and crosstalk are detailed in the related references. Bottom panel: Outline of signaling pathways and their effector mechanisms downstream of VEGF-A/VEGFR2. Numbers indicate phosphorylated tyrosines. PM, cellular plasma membrane.
The essential roles of these mediators in lymphangiogenesis is evident from studies on disruption of the respective genes in mice (eg, Vegf-C or Fox2c), which halts lymphatic system development. However, important clues have also been derived from expression studies. For example, the transmembrane guidance molecule, EPHRIN-B2 is coexpressed on the surface of LECs with its receptor EPHB4. This pathway was found to control lymphatic sprouting, as well as interaction of LECs and smooth muscle cells, and to play an essential role in remodeling of the lymphatic vasculature. Moreover, in the context of LECs (unlike in vascular ECs) ANG2 acts as an agonist of the TIE2 receptor, similarly to ANG1, a pathway crucial for lymphangiogenesis. Indeed, a germline ANG2 deletion in mice leads to lymphatic defects, which can be rescued by the expression of ANG1 (Augustin et al, 2009).
Proliferation, migration, and survival of LECs are regulated by a complex network of growth factors that straddle the processes of lymphangiogenesis and angiogenesis. Thus, VEGF-related factors, VEGF-C, and to some extent also VEGF-D stimulate lymphangiogenesis by interacting with VEGFR3/FLT-4. This interaction stimulates responses of LECs, but mainly when VEGF-C/D are present in their immature, proteolytically unprocessed forms. When proteolytically processed, these factors bind not only to VEGFR3 on LECs, but also to VEGFR2 on ECs, whereby they also stimulate angio-genesis. Moreover, VEGFR3 is also expressed on certain ECs under pathological conditions. Therefore, VEGF-C may stimulate angiogenesis also through this route (Tammela and Alitalo, 2010). Conversely, VEGF-A (often refered to simply as VEGF) may contribute to lymphangiogenesis through its interaction with VEGFR3 expressed on some LECs. VEGF-C also binds to Neuropilin-2 (NRP2), a semaphorin receptor, which interacts with VEGFR3 in a manner that is required for efficient lymphangiogenesis. VEGFR3 activation and lymphangiogenesis regulation also involve β1 and α9 integrins along with several other factors, such as Hepatocyte growth factor (HGF), Insulin-like growth factors 1 and 2 (IGF-1 and IGF-2), PDGF-B, and Fibroblast growth factors (FGFs). These interactions trigger formation of new lymphatic capillaries in a manner reminiscent of angiogenesis (Tammela and Alitalo, 2010).
11.3.6 Vasculogenic Mimicry
In some contexts nonendothelial cells may adopt endothelial-like phenotypes and line vascular channels (Hendrix et al, 2003). This process occurs in the normal placenta, where trophoblast epithelium enters the myometrium at sites of spiral arteries. This causes epithelial-to-endothelial transformation of trophoblastic cells, including the expression of several markers normally associated with ECs (CD31, CD144, Inte-grin αvβ3). Aberrant differentiation also occurs after subcutaneous injection of pluripotent embryonic stem (ES) cells into mice, and results in formation of aggressive teratomas, which contain blood vessels partially derived from ES cells (Li et al, 2009), albeit not always in high proportions (Rak, 2009). Similar transdifferentiation events were described in the bone marrow of patients with chronic myelogenous leukemia (CML), also in lymphoma, uveal melanoma, and glioblastoma (Hendrix et al, 2003; Ricci-Vitiani et al, 2010). The extent, significance, a nd molecular mechanisms of these processes are not fully understood, but several regulators have been implicated, including: tissue factor (TF), TF pathway inhibitor 2 (TFPI-2), phosphatidyl inositol 3 kinase (PI3K), focal adhesion kinase (FAK), MMPs, Ephrins, and Laminin chains (Dome et al, 2007).
11.3.7 Vascular Cooption
Cancer cells can exploit the preexisting tissue vasculature by growing around, and enveloping, the vessels (Holash et al, 1999). Various forms of this nonangiogenic process have been observed in highly vascular organs, such as lung and brain, as well as in melanoma and in liver metastases of colorectal cancer (Dome et al, 2007). Although cancer cells can grow and migrate along blood vessels, at least for a period of time, this interaction may trigger blood vessel regression and thrombosis (Brat and Van Meir, 2004), and is eventually followed by the onset of an angiogenic reaction (Holash et al, 1999).
11.4 MOLECULAR REGULATORS OF VASCULAR GROWTH
Formation of new vascular networks, whether in health or disease, is based on the ability of the constituent cells to participate in the web of intercellular communications. The mediators of these interactions can be broadly divided into several categories, including (a) specialized (“professional”) signaling effectors required for vascular homeostasis, (b) pleiotropic effectors endowed with angiogenesis-regulating activities (stimulators and inhibitors), along with other functions, and (c) other regulators that link angiogenesis to processes, such as hemostasis, bone marrow stimulation, neuronal growth, or immunity (Carmeliet and Jain, 2011). Although this molecular network contains redundancies, feedbacks and complex response patterns, at the heart of it are individual key molecules that can serve as potential targets for the anti-angiogenic therapy (Table 11–1 and Figs. 11–5 to 11–10). Key elements of this molecular circuitry are shown in Figure 11–5 and are discussed below.
TABLE 11–1 Key molecular regulators of tumor angiogenesis.
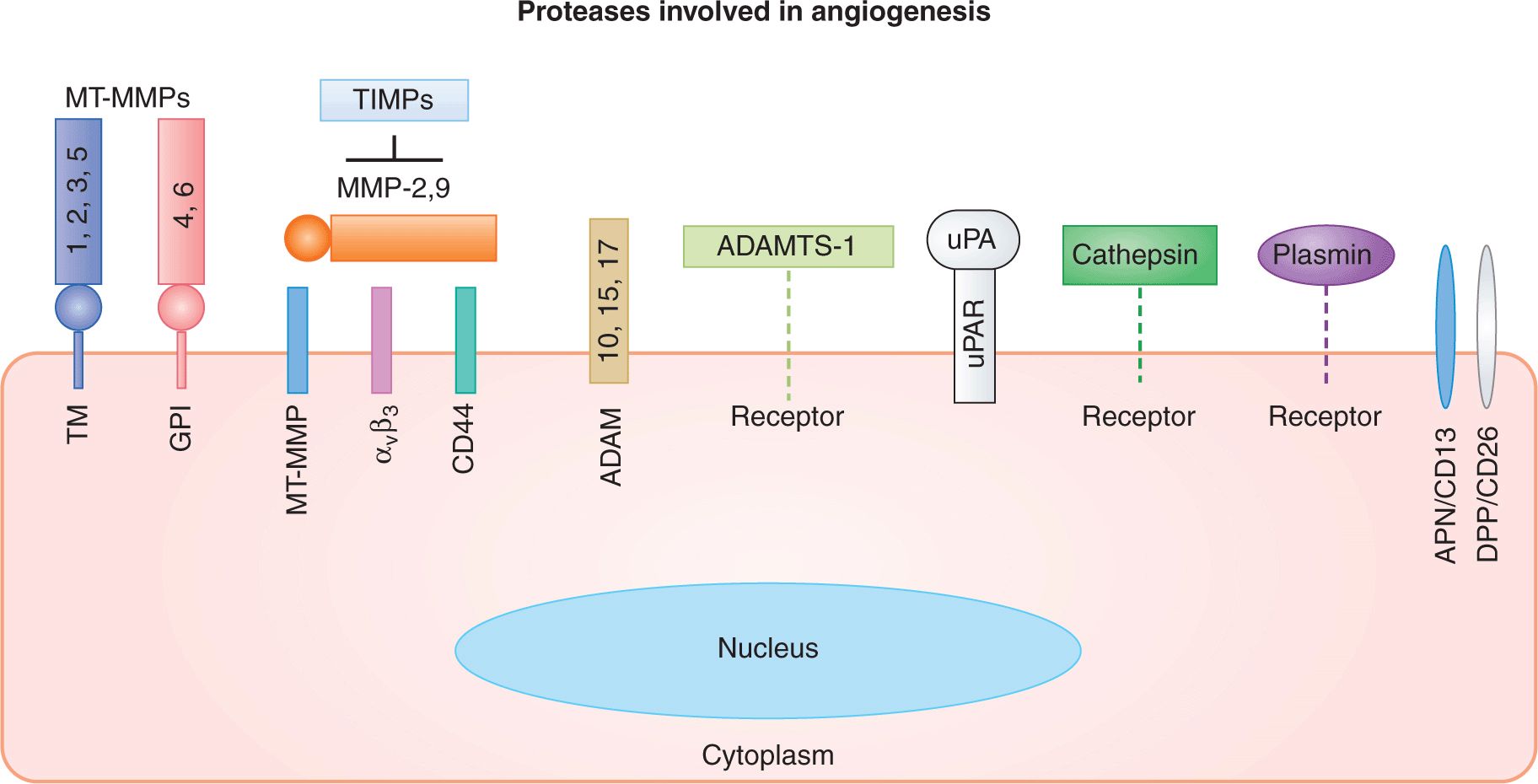
FIGURE 11–6 Proteolytic mechanisms involved in angiogenesis and endothelial cell function. Families of cellular and pericellular proteases involved in angiogenesis: ADAM, disintegrin and metalloproteinase domain-containing protein; ADAMTS, disintegrin and metalloproteinase domain-containing protein with thrombospondin motif; APN, aminopeptidase; DPP, diaminopeptidase; GPI, glycosylphosphatidylinositol anchor; MT-MMP, membrane-type matrix metalloproteinase; TIMPs, tissue inhibitors of metalloproteinases; TM, transmembrane region; uPA, urokinase plasminogen activator; uPAR, uPA receptor. (Adapted from van Hinsbergh et al, 2006.)
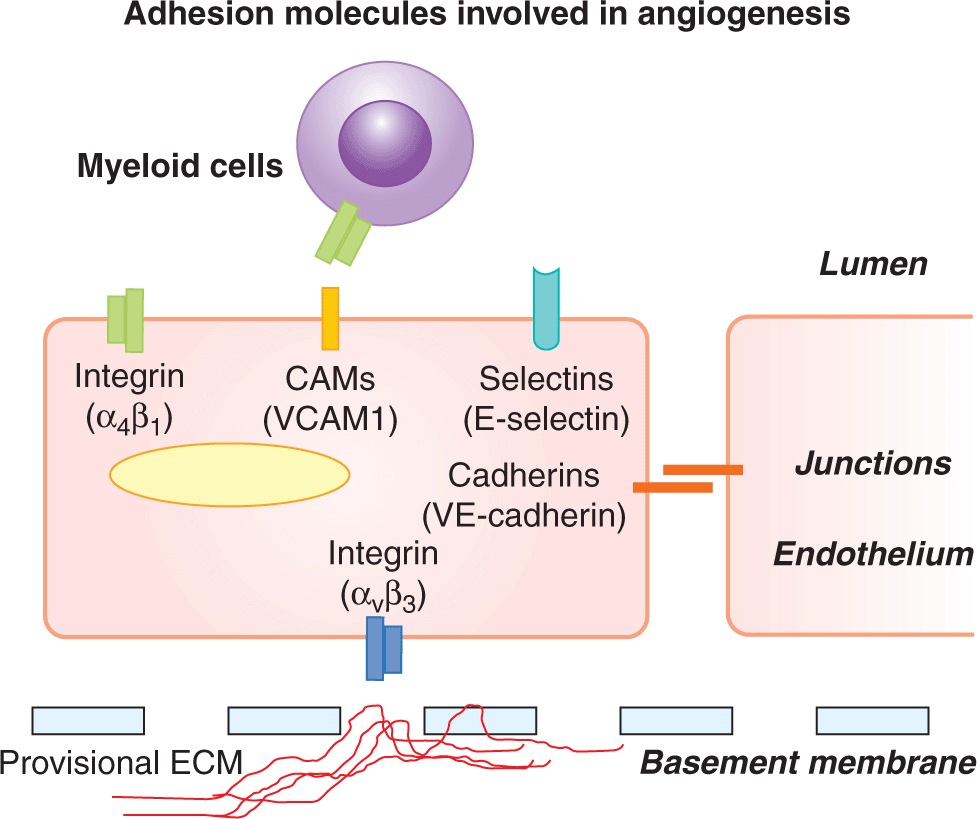
FIGURE 11–7 Adhesive mechanisms involved in angiogenesis and endothelial cell function. Endothelial cells interact homotypically through gap and tight junctional mechanisms, the latter involving lineage specific VE-Cadherin (CD144). Interactions with provisional ECM composed of proteolyzed collagen, fibrin, and vitronectin are mediated by αvβ3/β5 integrins. Cell adhesion molecules (CAMs), selectins, and integrins mediate interactions with circulating cells and platelets, play a role in mechanosensing, inflammation and permeability; see text.
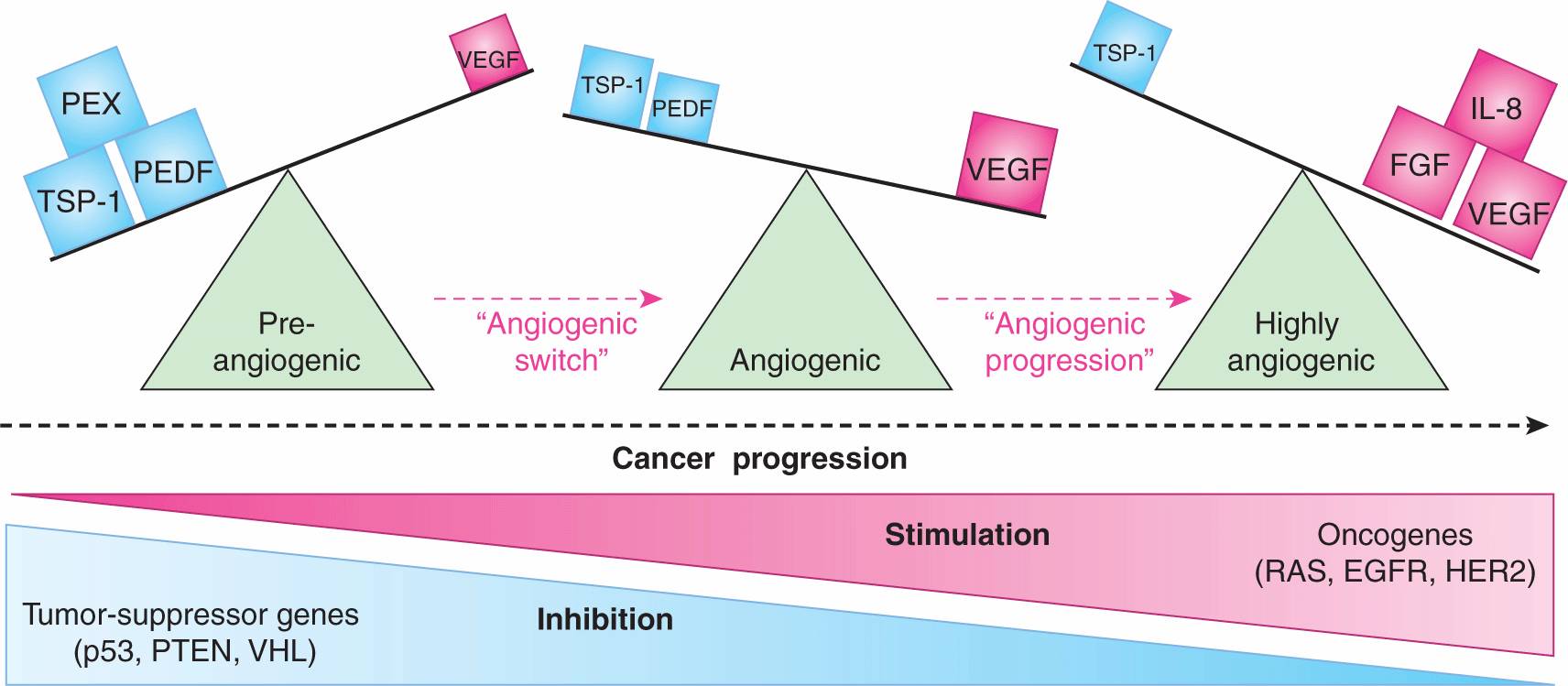
FIGURE 11–8 Angiogenic switch and progression during tumor development. Preangiogenic state in early tumor development is marked by the preponderance of angiogenesis inhibitors (TSP-1, PEDF, PEX), which override the effects of angiogenic stimulators (VEGF) expressed at low levels. Aggressive tumor growth is triggered by the change in balance between these factors, leading to the increase in the net stimulatory activity and the onset of angiogenesis (“angiogenic switch”). The underlying qualitative and quantitative changes in levels of angiogenesis stimulators and inhibitors are driven by tumor microenvironment (hypoxia, inflammation) and genetic progression of cancer cells (mutations in oncogenes and tumor suppressors). Continued escalation of these molecular changes with progressive disease may result in exuberant proangiogenic microenvironment with increasingly active and redundant stimulatory networks (angiogenesis progression), but excessive amounts of VEGF may be incompatible with robust angiogenesis. Abbreviations: PEDF, pigment epithelium derived factor (angiogenesis inhibitor); TSP-1, thrombospondin 1; PEX, non-catalytic fragment of MMP (all angiogenesis inhibitors); FGF, fibroblast growth factor; IL-8, interleukin 8; VEGF, vascular endothelial growth factor/VEGF-A (angiogenesis stimulators); see text.
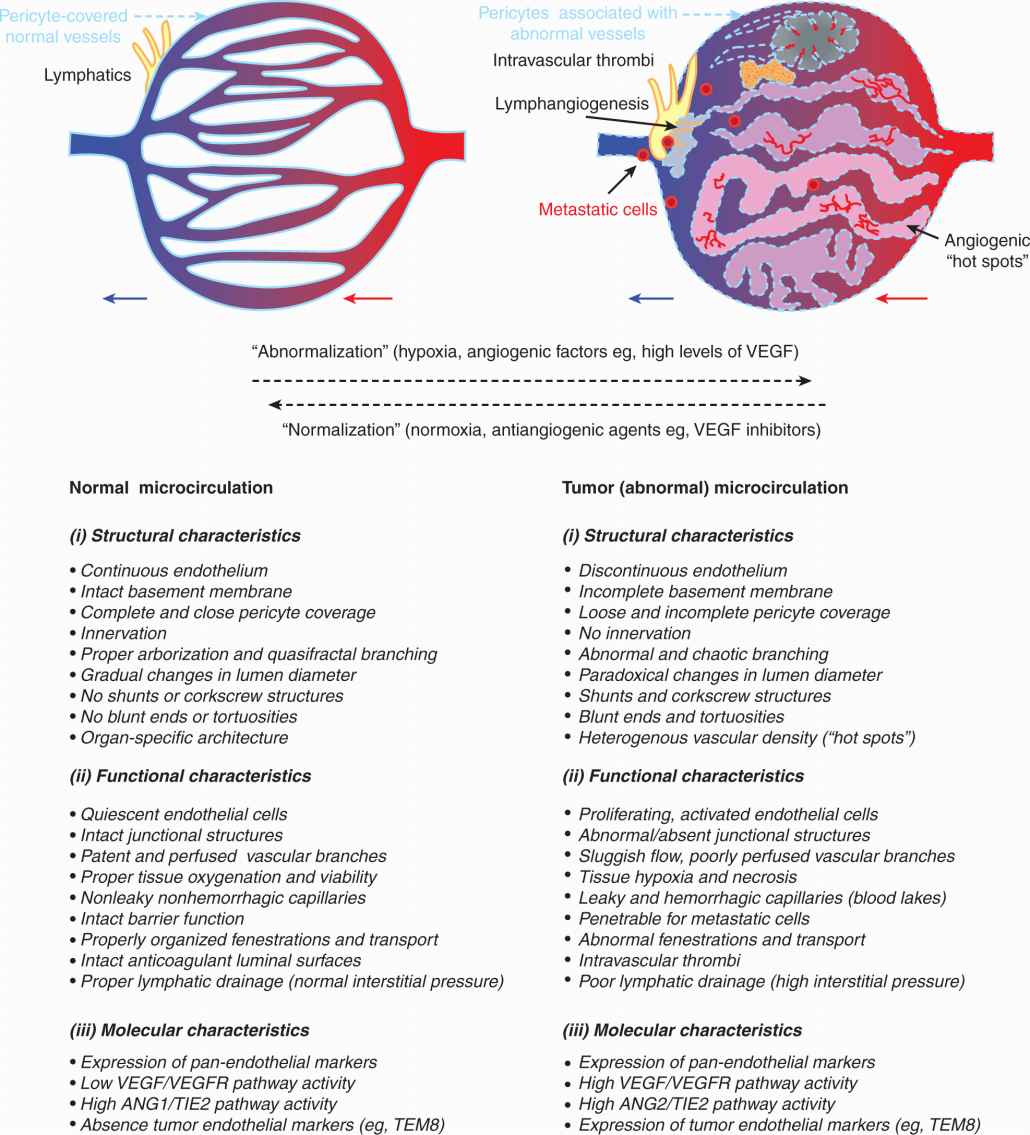
FIGURE 11–9 Tumor-related changes in vascular characteristics. Disorganized regulatory network in the tumor microenvironment leads to formation of blood vessels that are structurally, functionally, and molecularly abnormal (vessel “abnormalization”). Some of these abnormalities provide targets for antiangiogenic therapy that can selectively obliterate tumor blood vessels. Blood vessel-directed agents may either destroy blood vessels or restore, at least partially, some of their normal characteristics (vessel normalization); see text for details.
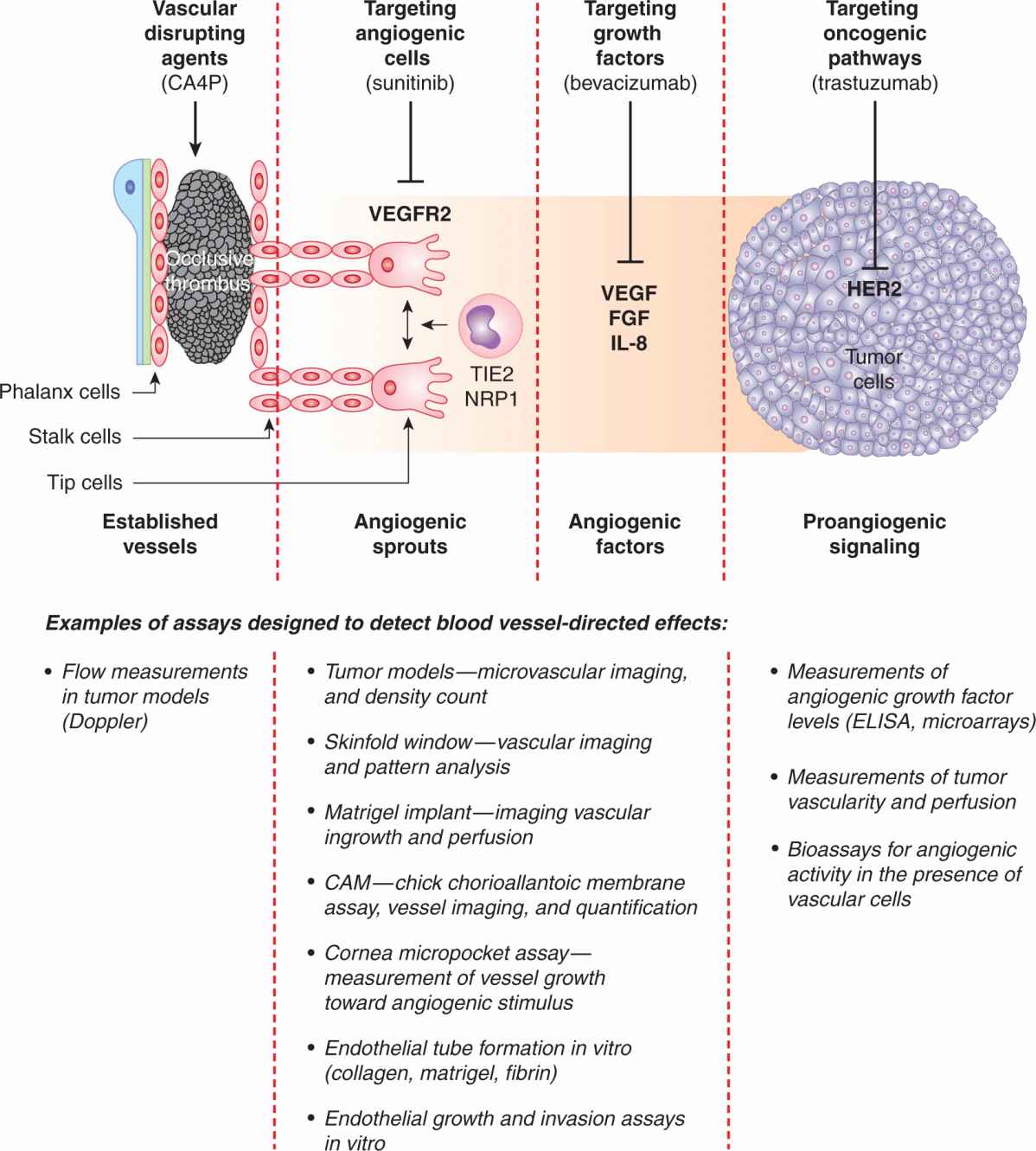
FIGURE 11–10 Some of the main strategies to target tumor vasculature. Development of the tumor microcirculation can be therapeutically opposed in several ways, including blocking mechanisms of the “angiogenic switch” through the use of antagonists of oncogenic pathways, such as trastuzumab, a neutralizing antibody that binds to HER-2 oncoprotein. Angiogenic factors, such as VEGF, can also be obliterated using neutralizing antibodies (eg, bevacizumab). Cells involved in angiogenic responses, namely, endothelial cells, PCs, and myeloid cells, can be prevented from responding to angiogenic growth factors by agents blocking their receptors, such as VEGFR2-directed drugs (eg, sunitinib or pazopanib). These cells can also be targeted using direct angiogenesis inhibitors (thrombospondin, tumstatin, integrin inhibitors, metronomic chemotherapy, and other agents). While these strategies generally prevent formation of new blood vessels, vascular disrupting agents (VDAs) obliterate the already established tumor vasculature by causing endothelial damage and thrombosis. Several assays have been developed to detect and measure the responses of the microcirculation to these respective insults (bottom panel); see text for details.
11.4.1 Vascular Endothelial Growth Factor Family
VEGF-A, also known as vascular permeability factor (VPF), is the key member of a larger family of related polypeptides, which also includes VEGF-B, VEGF-C, VEGF-D, VEGF-E, VEGF-F, and placenta growth factor (PlGF), all related to platelet-derived growth factor (PDGF) (Dvorak et al, 1995; Ferrara, 2005). These ligands form functional homo- and heterodimers and interact with at least 4 different receptors (VEGFRs), of which VEGFR1/FLT-1, VEGFR2/KDR/FLK-1), and VEGFR3/FLT-4 are signaling tyrosine kinases (RTKs), while Neuropilin 1 (NRP1) is a coreceptor (Ferrara, 2005). The relatively selective (albeit not exclusive) expression of VEGFRs by ECs (of blood vessels, lymphatics, and in tumors) allows VEGFs to control vascular processes in a potent and combinatorial manner (see Fig. 11–5). Thus, VEGFR1 binds VEGF-A, VEGF-B and PlGF, while VEGFR2 interacts mainly with VEGF-A (and VEGF-E), and VEGFR3 interacts preferentially with VEGF-C and VEGF-D. NRP1, the semaphorin receptor involved in neuronal guidance, is also expressed by ECs, where it acts as a coreceptor for VEGFR2. NRP1 binds only certain VEGF-A splice isoforms (eg, VEGF165) (Ferrara, 2005).
Germline deletion of each of the VEGF receptors leads to early embryonic lethality amidst vascular defects (Carmeliet and Jain, 2011), suggesting their essential role in vascular development and angiogenesis. However, the angiogenic responses elicited by exposure to the naturally occurring VEGF-A homodimers are mainly mediated mainly by VEGFR2. This involves robust phosphorylation and recruitment of intra-cellular signaling targets (see Fig. 11–5). Although VEGFR1 also binds VEGF-A with high affinity (10-fold greater than VEGFR2), the phosphorylation of this receptor in ECs is weak, and deletion of its kinase domain is relatively inconsequential for vascular development (Carmeliet and Jain, 2011). Indeed, VEGFR1 is often expressed as a splice variant composed of the soluble extracellular domain (sFlt-1), which acts as a natural VEGF antagonist (VEGF “sink”) (Carmeliet and Jain, 2011). However, VEGFR1 does possess an important regulatory function for macrophages, and certain cancer cells, in which it mediates migratory responses in response to VEGF (Carmeliet and Jain, 2011).
The crucial role of VEGF/VEGF-A is underscored by the unprecedented embryonic lethality and profound vascular defects in mouse embryos lacking even a single vegf allele (haploinsufficiency) (Carmeliet and Jain, 2011; Ferrara, 2005). Indeed, VEGF-A acts as a potent mitogen, motility, and survival factor for ECs, and a chemoattractant for their progenitors (Ferrara, 2005). These powerful influences are regulated by the organ- and context-specific alternative splicing of the VEGF-A transcript resulting in several protein isoforms. The main species generated during this process are designated according to the number of their constituent amino acids and include VEGF121 (121 amino acids), VEGF145, VEGF165, VEGF189, and VEGF206 (Ferrara, 2005). This splicing removes various sequences from within the region encoded by exons 6 and 7, while leaving the sequences corresponding to exons 1 to 5 and 8 intact.
Consequently, VEGF-A splice isoforms exhibit reduced heparin binding, increased solubility and diminished association with both cellular membranes and the ECM, depending on their size. Thus, the shortest VEGF121 isoform is highly soluble, while VEGF189 is mostly cell bound, and VEGF165 expresses intermediate properties, and the greatest angiogenic activity. The latter is a function of the more stable angiogenic gradient that VEGF165 (VEGF164 in the mouse) can form within the interstitial space in tissues. This gradient favors directional responses of tip cells and robust sprouting. In addition, unlike VEGF121, VEGF165 interacts with both VEGFR2 and NRP1, which is thought to contribute to more pronounced endothelial responses (Ferrara, 2005). Notably, the VEGF-A splicing process also results in expression of alternative ligands endowed with antagonistic (antiangiogenic) activities. For instance, one such variant, VEGF165b, interferes with angio-genesis, and exerts a modulating influence on responses of blood vessels to angiogenic growth factors involved in tissue remodeling and inflammation (Nowak et al, 2008).
The remaining members of the VEGF family play more restricted roles in vascular processes, and germline deletions of the respective genes do not have haploinsufficient consequences. VEGF-C/D control lymphangiogenesis, while VEGF-B provides additional survival protection to ECs. VEGF-E is a VEGF-like gene found in the genome of the Orf virus (Ferrara, 2005). In some instances, the effects of these factors become prominent only under pathological conditions (eg, in the case of PLGF). Finally, although VEGF-related factors were originally viewed as having mainly paracrine effects, VEGF-A was recently shown to be expressed at low, but functionally meaningful, levels by ECs themselves, which contributes to vascular homeostasis (Lee et al, 2007).
11.4.2 Platelet-Derived Growth Factor Family
This family of VEGF-related growth factors consists of 4 members: PDGF-A, PDGF-B, PDGF-C, and PDGF-D, the homo- or heterodimers of which interact preferentially with 1 of 2 main cellular receptor tyrosine kinases, namely, PDGFRα and PDGFRβ, (Andrae et al, 2008). While PDGFs play multiple roles in development, disease, and cancer, they are also central to vascular growth, especially blood vessel maturation. Thus, PDGF-BB homodimers are produced at high levels in endothelial tip cells and in phalanx cells of arteriogenic vessels, whereby they attract mural cells harboring PDGFRβ (Andrae et al, 2008). Other members of this family may also contribute to various angiogenic events indirectly, for example, by influencing the expression of angiogenesis-related genes (eg, VEGF) in cancer cells and fibroblasts (Dong et al, 2004). Indeed, recent studies suggest that these mechanisms may bypass the requirement for VEGF and contribute to tumor resistance to VEGF-directed therapies (Shojaei et al, 2009).
11.4.3 Prokineticins
This family of VEGF-unrelated growth factors has been found to induce VEGF-like effects in ECs, especially in endocrine organs. This includes growth, migration, permeability, fenestration (formation of trans-endothelial openings), and angio-genesis (LeCouter et al, 2002). These mediators consist of the endocrine gland VEGF/prokineticin 1 (EG-VEGF/PK1) and Bombina variegata-secreted protein 8/prokineticin 2 (Bv8/PK2), which both interact with their respective G-protein–coupled receptors (PK-R1 and PK-R2) (Ferrara, 2010). Although prokineticins appear to have a role in homeostasis of the microcirculation in endocrine glands, they are also found in cancer, where they may be produced by myeloid cells and contribute to VEGF-independent angiogenesis (Shojaei et al, 2009).
11.4.4 Angiopoietins and TIE Receptors
At least 3 related ligands known as angiopoietins (ANG1, -2, and -4) play diverse roles in endothelial cell survival, vascular development and maturation, as well as angiogenesis and lymphangiogenesis in humans (Ang3 is a mouse ortholog of ANG4) (Yancopoulos et al, 2000; Jones et al, 2001). Angiopoie-tins interact with TIE2/TEK receptor tyrosine kinase expressed preferentially (but not exclusively) on ECs, which also harbor a related orphan receptor known as TIE1 with poorly understood function. Nonetheless, both TIE1 and TIE2 receptors are essential for vascular development (Augustin et al, 2009). Their activity is controlled by the vascular endothelial receptor tyrosine phosphatase (VE-PTP) (Li et al, 2009). As mentioned earlier, the best understood function of the angiopoietin/TIE2 circuitry is in the regulation of EC survival, vascular permeability, and recruitment of mural cells. In this setting, ANG1 emanates from perivascular tissues and acts as the TIE2 agonist. In contrast, ANG2 is produced largely by ECs exposed to VEGF, and it blocks ANG1/TIE2 interaction, thereby destabilizing endothelial-PC contacts (see Fig. 11–5). Thus, in the presence of VEGF, the exposure to ANG2 promotes vascular sprouting, but when VEGF levels are low, ANG2 promotes vascular regression (Augustin et al, 2009 for review).
11.4.5 Notch Pathway
Cell-cell contact-dependent regulatory interactions often involve Notch receptors (NOTCH 1 to 4) and their 5 cell-associated ligands, including Jagged (JAG) 1, JAG 2, Delta-like 1, 3, and 4 (DLL1, DLL3, and DLL4) (Dufraine et al, 2008). NOTCH 1 and 4 are expressed in ECs (NOTCH 4 preferentially) and, along with DLL4, are required for proper vascular development. DLL4 is expressed in angiogenic tip cells and plays a role in maintaining their identity, whereas JAG 1 may be involved in modulating these effects and recruitment of additional tip cells. JAG 1 may also mediate direct interactions between endothelial and mural cells, or between tumor cells and the vasculature (Dufraine et al, 2008). Another Notch ligand, DLL1, is involved in vascular remodeling and arteriogenesis. Interactions with these ligands trigger proteolytic release of the intracellular domain of NOTCH (ICN), which is responsible for modulation of gene expression and cellular effects (Dufraine et al, 2008).
11.4.6 Ephrins and EPH Receptors
The definition of arterial and venous identity in the developing vascular system is largely attributed to the unique bidirectional signaling mechanism mediated by transmembrane ligands of the ephrin family, notably Ephrin B2 (on the arterial side) and their EPHB4 receptors (on the venous side; see Fig. 11–5). The expression of other Ephrins (eg, A1, A2, B1 and B3) and additional EPH receptors (EPHB2, EPHB3) is also observed in ECs, including in tumors. These Ephrins are implicated in various angiogenesis-related processes, such as endothelial–PC interactions, interactions of blood vessels with tumor cells, and cooperation with other angiogenic factors. The latter is exemplified by Ephrin B2-dependent regulation of VEGFR2 endocytosis and signaling (Wang et al, 2010).
11.4.7 Vascular Integrins, Cadherins, and Adhesion Molecules
Adhesion molecules connect ECs with their extraluminal, intercellular, and intraluminal surroundings (see Fig. 11–7). Thus, direct interaction between the abluminal surfaces of ECs and the extravascular ECM (basement membrane) is essential for survival, homeostasis, and angiogenic activity of these cells. Quiescent ECs are anchored to the permanent basement membrane, which is composed of laminin and collagen type IV. In contrast, angiogenic ECs are surrounded by provisional ECM containing fibrin, vitronectin, fibronectin, and partially proteolyzed collagens. These various interactions are mediated by a family of heterodimeric, transmembrane ECM receptors, known as integrins (each composed of α and β subunits), which recognize specific motifs within their target ECM proteins (eg, RGD peptides).
Integrins are viewed as functional hubs that localize and regulate the activities of other angiogenic effectors, including VEGFR2, other receptors, MMPs, angiopoietins, and intracellular signal transducing kinases (PKB/AKT, FAK, ILK, SRC). Growth factors upregulate the expression of several Integrins on the surface of ECs in blood vessels (αvβ3, αvβ5, α1β1, α2β1, α4β1, α5β1) and lymphatics (α1β1, α2β1, α4β1, α9β1)(Avraamides et al, 2008). Although acute pharmacological disruption of the adhesive function of certain integrins (αvβ3) resulted in antiangiogenic effects (Cheresh and Stupack, 2002), the corresponding gene targeting studies suggested a more complex involvement. This is illustrated by the enhanced, rather than reduced, tumor angiogenesis in β3/β5-deficient mice (Reynolds et al, 2002). Indeed, other mutations of vascular integrins may either reduce (β3 mutant, α1β1), or increase (α2β1) adult angiogenesis (Avraamides et al, 2008).
In contrast to the outward-oriented adhesion that is mediated by endothelial integrins, vascular endothelial cadherin (VE-cadherin/CD144) mediates formation of intercellular junctions between ECs within the vascular tube. This contact contributes to the unique properties of the endothelial lining (barrier function, restricted permeability, homotypic adhesion) (Nyqvist et al, 2008). VE-cadherin is selectively expressed by the cells of endothelial lineage and serves as their genetic marker. Importantly, gene targeting studies revealed that VE-cadherin is essential for vascular development, and for the functionality of some of its key regulators, such as VEGFR2 (Carmeliet and Jain, 2011). N-cadherin is also expressed by ECs, albeit not as selectively, and is required for vascular integrity and developmental angiogenesis. Cadherins bind to other cadherins on the adjacent cells and orchestrate formation of physical contacts, referred to as adherens junctions. These structures along with tight junctions (involving claudins) influence the functional integration of the endothelial lining.
Interactions between the endothelium and circulating immune, myeloid, inflammatory, and progenitor cells, and platelets are mediated by a diverse class of intraluminal adhesion molecules, including selectins (eg, E-selectin), integrins (α4β1/VLA4), and members of the immunoglobulin family of cell adhesion molecules (CAMs, eg, ICAM-1/2 and VCAM1), all of which play different roles in angiogenesis (Francavilla et al, 2009).
11.4.8 Angiogenesis-Regulating Proteases
During blood vessel formation, the dissolution of the endothelial basement membrane, liberation of progenitor cells from the bone marrow, release of VEGF and other growth factors from their ECM stores, and the activation/modulation of the coagulation system, all involve various classes of proteases and their endogenous inhibitors (Kalluri and Zeisberg, 2006; van Hinsbergh et al, 2006). Of particular note are matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs 1 to 4), often expressed in the tumor microenvironment (see Fig. 11–6). These molecules participate in ECM breakdown, generation of angiogenesis-regulating protein fragments, regulation of tumor and endothelial cell invasion, and may also act as ligands of cellular receptors (Kessenbrock et al, 2010). The main MMPs involved in tumor angiogenesis are MMP-1, MMP-2, MMP-9, and MMP-14. Their role is illustrated by the impairment of tumor neovascularization in mice with disrupted MMP9 gene expression (Kessenbrock et al, 2010). Other proteases involved in angiogenesis include cathepsins, coagulation factor VIIa, thrombin (IIa), urokinase-type plasminogen activator (uPA), as well as members of the disintegrin and metalloproteinase domain (ADAM) and thrombospondin motif-containing (ADAMTS) families of proteases (van Hinsbergh et al, 2006).
11.4.9 Angiogenesis Stimulators and Inhibitors
The mechanisms described above are required for the execution of one or more of the angiogenic programs, but they are not necessarily the only or initial triggers of tumor angiogenesis. In cancer and other angiogenesis-dependent diseases, vascular growth may be initiated by a global shift in expression of several molecules, and the related cumulative change in the regulatory environment, the constituents of which are broadly classified as angiogenesis stimulators and inhibitors (see Table 11–1). It is believed that the onset of angiogenesis (“angiogenic switch,” see Fig. 11–8) is triggered when the balance between these opposing influences is tilted beyond a certain discrete threshold, and in favor of stimulators (Folkman and Kalluri, 2003). Although this transition may appear as a binary event, the biological mechanisms involved can be complex, discontinuous, incremental, or oscillatory. The angiogenic threshold and the magnitude of endothelial cell responses may also differ depending on the genetic background and several other factors (Rohan et al, 2000). Furthermore, the composition of angiogenic factors may change over time during tumor progression (see Fig. 11–8), leading to a series of transitions rather than a single switch (Folkman and Kalluri, 2003).
Stimulators of angiogenesis include factors acting directly on ECs, such as VEGF, PlGF, FGF1/2, HGF, or interleukin 8 (IL-8). Similar effects can also result from indirect actions of their potent inducers, for example, transforming growth factors alpha or beta (TGFα, TGFβ) and several other cytokines and chemokines (eg, pleiotrophin). Some of these factors may act by recruitment of inflammatory cells (eg, IL-6), or bone marrow progenitor cells (eg, VEGF or stromal derived factor 1 [SDF1]), or through concomitant stimulation of both endothelial and inflammatory cells (eg, IL-8) (Kerbel, 2008).
The endogenous angiogenesis inhibitors are thought to maintain blood vessels in their quiescent state, or limit the magnitude of angiogenic responses, both locally and systemically (Folkman and Kalluri, 2003). Angiogenesis inhibitors belong to different classes of molecules, including certain ECM proteins (thrombospondins 1 and 2) (Bouck et al, 1996), or their proteolytic fragments (endostatin, tumstatin, arresten), fragments of enzymes and zymogens (angiostatin, PEX domain), fragments of coagulation-related chemokines (platelet factor 4 [PF4]), and of hormones (prolactin 16-kDa fragment), along with certain cytokines (interferons α, β, and γ) (Folkman and Kalluri, 2003; see Table 11–1).
11.4.10 Coagulation System and Platelets as Regulators of Tumor Angiogenesis
The discovery of VEGF in 1983 (Senger et al, 1983), and its role as a vascular permeability factor (VPF), originally linked angiogenesis to the coagulation system, especially through the observation of the associated extravascular leakage of plasma and formation of proangiogenic fibrin matrix (Dvorak et al, 1995). Although tumor angiogenesis was found to occur also in the absence of fibrinogen and fibrin, proangiogenic activities can be ascribed to several effectors of the coagulation system, such as TF/thromboplastin, thrombin and several others (Rickles et al, 2003). These factors may act directly on ECs, or stimulate the expression of VEGF, IL-8, and other angiogenic proteins by cancer cells and stroma. An important role is attributed to circulating blood platelets (Pinedo et al, 1998), which may serve as reservoirs and carriers of angiogenic factors (eg, plasma VEGF) and inhibitors (PF4, TSP-1). Indeed, platelets appear to take up, accumulate and segregate such factors in their granules and may selectively release them at sites of angiogenesis (Klement et al, 2009).
In addition to simple molecular signals (eg, soluble VEGF), ECs and other elements of the tumor microenvironment may receive and emit more complex (multimolecular) messages, encapsulated in cell membrane-derived structures known as microvesicles, microparticles, or exosomes. Microvesicles may deliver angiogenic growth factors (VEGF, FGF), inflammatory cytokines (IL-1), enzymes (MMPs), enzyme inducers (EMMPRIN), and other mediators, including messenger RNA (mRNA), microRNA, and DNA. Interestingly, microvesicles derived from cancer cells may also contain active and phosphorylated oncoproteins that may enter and reprogram ECs (eg, trigger autocrine production of VEGF) thereby possibly contributing to tumor angiogenesis (Al-Nedawi et al, 2009).
11.5 TUMOR ANGIOGENESIS
11.5.1 The Onset and Progression of Angiogenesis in Cancer
Cancer cells are dependent on their proximity to vascular networks for their growth, survival, and metastatic dissemination (Folkman and Kalluri, 2003). Sometimes these requirements are satisfied by vascular cooption, invasion of the preexisting vessels, postnatal vasculogenesis, or vascular mimicry as described earlier. However, in most human cancers the vascular dependence of tumor cells is met through the onset of angiogenesis, that is, tumor formation and progression can be regarded as angiogenesis dependent (Folkman and Kalluri, 2003). In some cases VEGF expression can be rate limiting for these events, which therefore can be viewed as VEGF dependent. Because of the central role of VEGF in developmental angiogenesis, this concept has undergone considerable (excessive) generalization, and has influenced design of antiangiogenic agents and clinical trials to evaluate them. Thus, the majority of clinically used antiangiogenic drugs are presently directed at VEGF or VEGFRs (Table 11–2) (Ferrara, 2005).
TABLE 11–2 Examples of blood vessel-targeting agents developed to treat cancer.
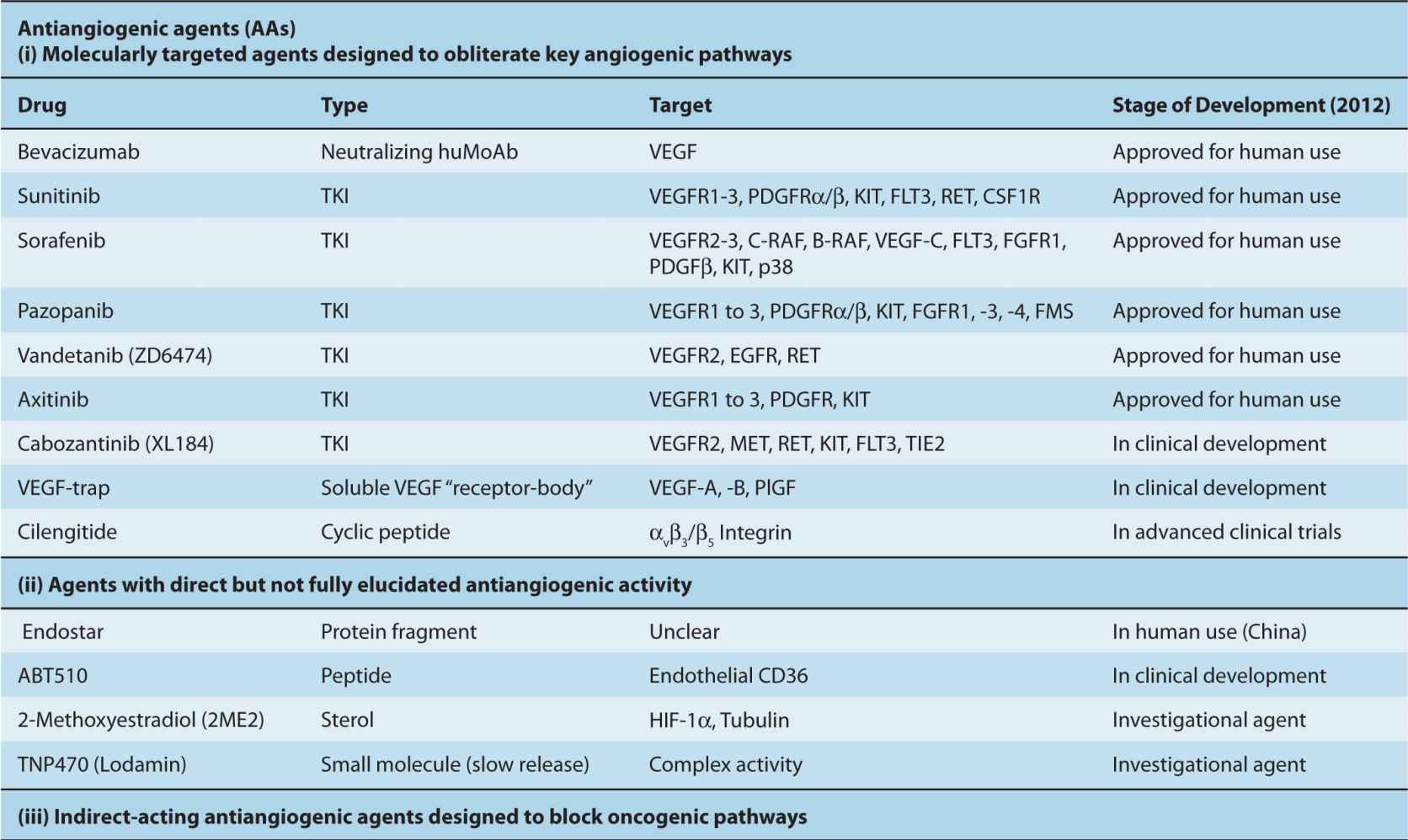
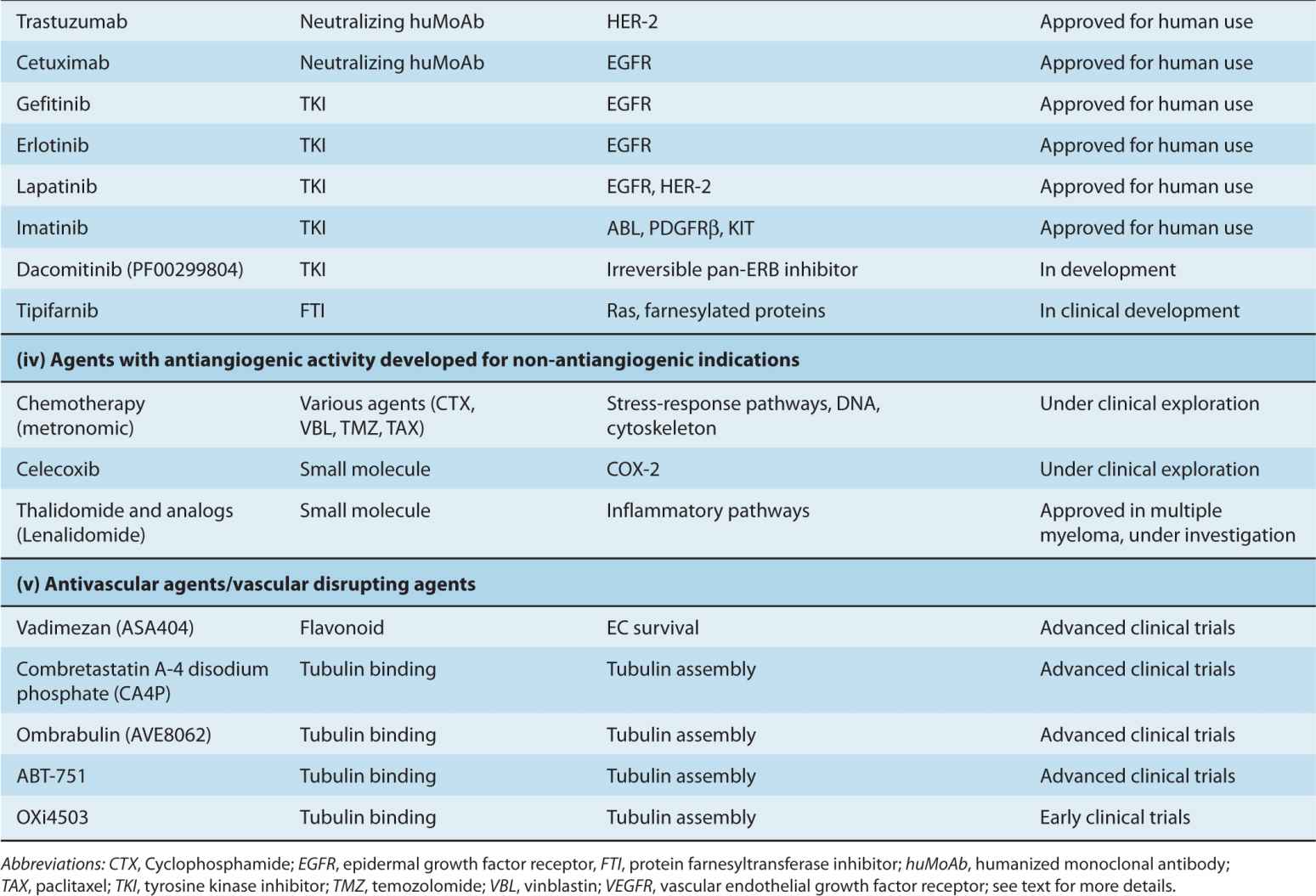
The processes of vascular maturation are also disorganized in the context of cancer, which results in an incomplete and heterogeneous PC coverage (McDonald and Choyke, 2003). These defects may impact the susceptibility of tumor blood vessels to VEGF-directed therapies, as ECs devoid of PC contacts are more dependent on VEGF for survival (Ferrara, 2005). When such therapies exhaust their antiangiogenic impact a preponderance of more mature (resistant) and well-perfused tumor blood vessels is often observed. Incomplete blood vessel suppression combined with a shift toward better perfused and more mature microcirculation is often referred to as vessel normalization (Jain, 2001). Although this process may diminish the impact of VEGF inhibitors, it might also be exploited therapeutically, for example, by taking advantage of the increased delivery of anticancer chemotherapy agents to cancer cells that are better supplied with blood due to a more functional (“normalized”) vascular network (Jain, 2001). Moreover, PCs themselves may serve as therapeutic targets in cancer, especially in situations where they support a significant proportion of tumor blood vessels (Carmeliet and Jain, 2011).
11.5.2 Mechanisms Triggering Tumor Neovascularization
The onset of tumor angiogenesis is often viewed as an early event in the course of disease progression, and one that permits the emerging tumor mass to expand beyond the size of 1 to 2 mm in diameter. The “preangiogenic” phase of tumor growth is defined by the limits of oxygen and macromolecule diffusion from the preexisting host vasculature (see Fig. 11–8). Other factors that influence the limits of preangiogenic tumor growth include the ability of cancer cells to tolerate hypoxia and metabolic deprivation (Folkman and Kalluri, 2003; see Chap. 12, Sec. 12.3), as well as the degree to which such cells can utilize vascular cooption and other alternative, nonangiogenic mechanisms of gaining an access to vascular networks (Holash et al, 1999). Beyond the boundaries of perivascular diffusion, cancer cells either undergo growth arrest, or enter a state of dynamic dormancy, defined by a balance between growth and cell death, without a net increase in the tumor mass (Folkman and Kalluri, 2003). Such exposure may eventually drive the onset of angiogenesis due to secondary changes in phenotype.
As mentioned earlier, the onset of angiogenesis (“angiogenic switch”; see Fig. 11–8) is often attributed to the net increase in the activity of angiogenesis-stimulating influences over those of inhibitors (Folkman and Kalluri, 2003). Cancer cells play a central and causative role in these events, and their role may be either direct (as producers of angiogenic factors), or more indirect, for example related to their ability to cause a hypoxic shift in the tissue microenvironment, recruitment/activation of proangiogenic stromal cells, and/or induction of inflammatory responses (Rak, 2009). Even within the same lesion the various subsets of cancer cells may differ in their proangiogenic activity. In some instances, this capacity appears to be particularly pronounced in the case of tumor-initiating cells (TICs), or cancer stem cells (CSCs), a notion particularly well studied in the case of brain tumors (Gilbertson and Rich, 2007). The major underlying trigger of a cancer cell-related angiogenic phenotype lies with the interplay between the effects of intracellular oncogenic pathways and responses to the extracellular tumor microenvironment, especially hypoxia and inflammation (see Chap. 12).
11.5.3 Hypoxia as a Trigger of Tumor Angiogenesis
Hypoxia acts as the primary physiological trigger of blood vessel formation, for example, during development, ischemia, and wound healing. Hypoxia is usually defined as tissue oxygen level below 10 to 15 mm Hg (see Chap. 12, Sec. 12.2), and may originate from several changes, such as increased oxygen consumption, low capillary density, poor blood perfusion, and the presence of vascular occlusion. Hypoxic regions (and areas of necrosis) are relatively common in advanced solid tumors, and their presence is usually associated with poor prognosis. Poor oxygenation evokes cellular responses involving the expression of genes that control intrinsic coping mechanisms, such as glycolytic metabolism, cellular quiescence, cell survival, and DNA repair, as well as those responsible for extracellular effects such as erythropoiesis and angiogenesis (Bristow and Hill, 2008).
The best described amongst the hypoxia response pathways is the activation of hypoxia-inducible transcription factors 1 and 2 (HIF1/2; see Chap. 12, Sec. 12.2.3). Several angiogenesis-related genes are targets of the HIF pathway, including VEGF, SDF1, ANG2, PlGF, PDGF-B, stem cell factor (SCF), and endothelial VEGFR1 (Rey and Semenza, 2010). Hypoxia also leads to downregulation of some angiogenesis inhibitors, notably thrombospondin 1 (TSP1) (Laderoute et al, 2000). HIF-mediated transcription is also involved in angiogenic responses of ECs themselves and contributes to vascular permeability and vessel ‘abnormalization’ (Fig. 11–9), followed by increased shedding of metastatic cancer cells into the blood stream (Carmeliet and Jain, 2011). Hypoxia provokes tumor cell invasiveness (Rey and Semenza, 2010) and HIF has been proposed as a factor that maintains the cancer stem cell population (Hill et al, 2009). Several other transcription factors may contribute to proangiogenic responses under hypoxia, including: nuclear factor kappa B (NF-κB), NF-IL6, MTF-1, and EGR-1.
Hypoxia triggers both angiogenesis (locally) and arteriogenesis (remotely), but the nature of these two events is fundamentally different (Carmeliet and Jain, 2011). Hypoxia serves as a potent inducer of local VEGF production (along with other factors), which in normal tissues acts as a self-limiting growth stimulus for capillaries. Angiogenesis causes an increase in blood vessel numbers, improves perfusion and tissue reoxygenation, which causes cessation of the hypoxic stimulus. Arteriogenesis occurs upstream of these events and outside of the boundaries of the hypoxic tissue, and ensures the parallel enlargement of feeding vessels through the effects of monocytes and nitric oxide (NO) (Carmeliet and Jain, 2011).
11.5.4 Tumor Vasculogenesis
Vasculogenesis involves incorporation of circulating EPCs into the capillary wall of the emerging tumor vasculature (Rafii et al, 2002). Although the nature and extent of this process is somewhat controversial, the presence of circulating cells with EPC-like characteristics in tumor-bearing animals and in cancer patients is well established, as is the ability of such cells to populate certain vascular structures (Asahara et al, 1997; Bertolini et al, 2010). Moreover, experiments involving chimeric animals harboring genetically tagged bone marrow suggest that EPCs may contribute to as much as 50%, or (more frequently) to as little as 0.01% of tumor blood vessels (Ahn and Brown, 2009). Similarly, in cancer patients who have previously undergone a gender-incompatible bone marrow transplant, donor-related EPCs (containing chromosomes or genes that distinguish them from the recipient) were found to contribute to only a minor fraction (approximately 5%) of the tumor vasculature (Peters et al, 2005).
The incorporation of EPC-like cells into tumor blood vessels may be stimulated by cancer therapy. For example, when acute administration of cytoreductive, or antivascular agents provokes emission of bone marrow activating cytokines, EPC-like cells are found to emerge within the circulation and home to the tumor mass in relatively large numbers (Shaked et al, 2006). It is proposed that this is a result of the combined action of circulating VEGF, granulocyte colony-stimulating factor (G-CSF), SDF1, and adhesion mediated by α4 integrin. In this manner EPC-like cells may contribute to tumor revascularization and diminished antitumor effect of antivascular agents (Shaked et al, 2006). An important role in orchestrating the EPC recruitment is attributed to the ID1 transcriptional repressor (Rafii et al, 2002).
11.5.5 Proangiogenic Effects of Oncogenic Pathways
Cancer cells often express strongly proangiogenic properties (eg, overproduction of VEGF), even without exposure to hypoxia (Rak, 2009). This intrinsic and constitutive proangiogenic phenotype is triggered by the expression of several dominant oncogenes, examples of which include H-Ras, K-Ras, Src, Myc, EGFR, and HER2 (Rak, 2009). In addition, the loss-of-function mutations affecting tumor-suppressor genes, such as p53, PTEN, p16′NK4a, and VHL, also exert potent angiogenic effects, often in a cumulative manner (Bouck et al, 1996; Rak, 2009). These changes are executed through the pleiotropic impact these genetic events have on the cellular angiogenic transcriptome and proteome (angiome), via activation of kinase cascades, transcription factors and microRNA networks, such as the miR-17-92 cluster (Dews et al, 2006).
The most studied angiogenic effects of oncogenic mutations include upregulation of the key vascular stimulators—VEGF, FGF, IL-6, IL-8, or ANG1—coupled with downregulation of angiogenesis inhibitors, especially TSP-1, endostatin, tumstatin, and pigment epithelium-derived factor (PEDF) (Bouck et al, 1996; Rak, 2009). However, oncogene-regulated mediators of angiogenesis also include phospholipids, proteolytic enzymes, and microvesicles containing multiple angiogenic proteins (Al-Nedawi et al, 2009).
Activated oncogenic pathways often mimic or exacerbate cellular responses to hypoxia. For example, the extremely high levels of VEGF, florid angiogenesis, and hypervascularity observed in sporadic clear cell renal cell carcinoma (CCRCC) are largely attributed to the high frequency (70%) of the loss-of-function mutations affecting the VHL gene (Kaelin Jr, 2008). The resulting activation of HIF-dependent responses (see Chap. 12) drives transcription of VEGF and other vascular changes, even in the absence of overt oxygen deprivation. Similarly, while VEGF expression is upregulated by hypoxia in numerous types of nontransformed cells, the expression of RAS and other oncogenes in cancer cells can cause a marked amplification of these responses, leading to more exuberant production of VEGF (Mazure et al, 1996).
11.5.6 Inflammation
Tumor angiogenesis is profoundly affected by inflammatory cells and their soluble mediators. The sources of inflammatory reaction in cancer can be either extrinsic (infection), intrinsic (oncogenic mutations), immunological, or microenvironmental (Mantovani et al, 2008). These processes trigger recruitment of different subsets of inflammatory cells, especially M2-polarized (tumor-promoting) macrophages and other types of myeloid cells (see Fig. 11–2). There is also a growing interest in TIE2-expressing macrophages, mast cells (MCs), and granulocytes, many of which produce proteolytic enzymes, proangiogenic growth factors, cytokines, and chemokines (VEGF-A, Bv8, IL-8) (Schmid and Varner, 2010). VEGFA exerts promigratory effects on macrophages, but inhibits antitumor immune responses, thereby tilting the regulatory balance toward tumor-promoting processes. Both tumor cells and stroma may contribute to these events through the activation of abnormal cellular “defense” mechanisms orchestrated by transcription factors (eg, NF-κB, HIF, and STAT [signal transducers and activators of transcription]), in response to the hostile tumor microenvironment (Mantovani et al, 2008).
11.5.7 Angiogenic Activation of Tumor Stroma
The presence of host cells (stroma) within the tumor mass is central to the onset of angiogenesis. Stromal cells likely contribute to endothelial cell quiescence in normal tissues by producing architectural constraints and angiogenesis inhibitors. In cancer, stromal cells may undergo functional changes described as “activation,” or may even sustain genetic mutations (Hill et al, 2005). The sources of cancer-associated fibro-blasts are likely diverse, ranging from cooption of resident connective tissue cells, their phenotypic reprogramming, changes in ECs, eg, endothelial-to-mesenchymal transition (EnMT), or analogous processes involving cancer cells, that is, epithelial-to-mesenchymal transition (EMT) (Kalluri and Zeisberg, 2006). Cancer-associated fibroblasts have long been known to exhibit elevated production of VEGF-A, and may also produce other angiogenic factors. For example, production of PDGF-C by these cells was found to contribute to the resistance of tumors to therapies directed at VEGF-A. Hypoxia, paracrine interactions, activation of the coagulation system, inflammation, and oncogenic lesions may all contribute to the recruitment and stimulation of the angiogenic tumor stroma (Dvorak, 1986).
11.6 MODIFIERS OF TUMOR ANGIOGENESIS
Although most tumors express various elements of the angiogenic phenotype, the dynamics of the related vascular responses is often context dependent. This is defined by the tumor type (grade/molecular profile), or by host-dependent influences. The latter may be a function of individual characteristics of cancer patients, for example, their genetic background, age, metabolic conditions (eg, obesity, diabetes, atherosclerosis), and other comorbidities, such as thrombosis, postsurgical responses, constitutive levels of circulating growth factors, hormones, and other regulators. These host-related factors may impact various aspects of angiogenesis, such as microvessel density, endothelial cell proliferation, vascular branching patterns, and the numbers of recruited bone marrow cells at the tumor site.
11.6.1 The Impact of Genetic Background on Tumor Angiogenesis
A similar angiogenic stimulus may evoke angiogenic responses of differential nature and magnitude because of relatively subtle changes in the host genetic background. For example, different strains of mice exhibit inherently differential angiogenic reactions in response to stimulation with the same recombinant angiogenic growth factors (Rohan et al, 2000). Tumor angiogenesis was found to be constitutively altered in mice deficient for ID1 transcriptional repressor, β1 Integrins, β3/β5 Integrins, and other factors (Lyden et al, 1999; Reynolds et al, 2002). Similarly, mice expressing different levels of VEGF-A (eg, due to introduction of a hypomorphic VEGF allele) exhibit alterations in angiogenesis, including in association with cancer (Sung et al, 2010). Polymorphisms of the VEGF gene in humans regulate both the angiogenic activity and responses to antiangiogenic agents, for example, in breast cancer patients administered bevacizumab (see Sec. 11.7.1.3; Schneider et al, 2009). Supernumerary chromosome 21, which is associated with Down’s syndrome, impedes VEGF production and angiogenesis through the action of DSCR1 and DYRK1a genes (Reynolds et al, 2010). Moreover, otherwise silent heterozygosity of host cells for certain tumor-suppressor genes may influence regulation of angiogenesis and tumor progression. For example, in the murine model of neurofibromatosis where host tissues have the NF1+/– genotype, the related increase in proangiogenic mast cell recruitment is required for the efficient growth of NF1–/– tumors (Yang et al, 2008). Similar influences may also exist in cancer patients.
11.6.2 Modulation of Tumor Angiogenesis by Vascular Ageing and Comorbidities
Although cancers are more prevalent in adults and the elderly then in children, they may occur at any age, and the age of onset can influence tumor angiogenesis (Reed and Edelberg, 2004). This is exemplified not only by the uniqueness of malignancies affecting children, adolescents, and young adults (including some of their vascular phenotypes), but also by the age-related differences in more common cancers. For example, the age of patients affected by CCRCC may vary by up to 5 decades. During this time, the vascular system undergoes profound changes, including alterations in the state of endothelial, bone marrow, inflammatory, and other cellular compartments. Ageing impacts responses to hypoxia, impedes production of angiogenic growth factors and efficiency of vascular repair, it increases procoagulant tendencies and leads to cardiovascular decline. Age-related alterations in the vasculature may influence the responses to antiangiogenic therapies, and can be further compounded by age-related comorbidities affecting the macro- and microvasculature, eg, atherosclerosis (Rak, 2009).
11.6.3 Properties of the Tumor Microcirculation
Tumor blood vessels exhibit high levels of architectural, cellular, and molecular heterogeneity, in comparison to normal tissues (Jain, 2001). These anomalies include loss of proper arborization (hierarchy), unusual patterns of branching (trifurcation), paradoxical lumen dilatations, formation of corkscrew and blind ended structures, and abnormal vascular connections (shunts). Tumor blood vessels may contain capillaries with either incomplete or hypertrophic endothelial cell lining (eg, in brain tumors), which may fold into intraluminal projections and septa, and interact poorly with the surrounding PCs (McDonald and Choyke, 2003). Perivascular micro-hemorrhages and spaces filled with blood (blood lakes) are common in tumors along with vascular occlusion and regression (Holash et al, 1999). Regional abnormalities of the vessel wall often result in the leakage of plasma, macromolecules, and clotting factors into the extravascular space. This leads to extravascular activation of the coagulation system and deposition of crosslinked fibrin, as well as formation of intravascular occlusive thrombi (Brat and Van Meir, 2004). A combined effect of these changes amounts to sluggish, intermittent, often bidirectional and inefficient blood flow, poor tissue perfusion, hypoxia, and necrosis (Folkman and Kalluri, 2003).
Structural abnormalities of tumor blood vessels are associated with a shift in the EC phenotype. Thus, tumor-associated ECs may exhibit elevated mitotic indices (Denekamp, 1982), changes in cell shape, and regional heterogeneity in the expression of molecular markers. They express abnormal levels of various proteins and phospholipids (eg, exposed phosphatidylserine) (Neri and Bicknell, 2005). Some tumor ECs overexpress proangiogenic receptors (eg, VEGFR2 or TIE2), that stimulate angiogenic activity. Molecular profiling of ECs isolated from blood vessels of certain human and murine tumors has also revealed the expression of molecules normally absent in the vasculature, and often referred to as tumor endothelial markers (TEMs) (Seaman et al, 2007).
Tumor ECs may also exhibit genetic changes, for example, abnormal ploidy (Hida et al, 2004), marker chromosomes, or expression of mutant oncogenes, suggesting that some tumor cells (especially CSCs) may contribute to the endothelial lining (Ricci-Vitiani et al, 2010). The unique molecular make up of tumor ECs can be used to guide the development of blood vessel-directed anticancer therapies (Neri and Bicknell, 2005).
11.6.4 Tumor Lymphangiogenesis
Solid tumors often exhibit high interstitial fluid pressure indicative of poor lymphatic drainage (see also Chap. 12, Sec. 12.2.4). This property was long regarded as an indication that lymphatics are absent in growing tumor masses, but more recent studies provided evidence to the contrary. For instance, lymphangiogenesis and expression of lymphangiogenic growth factors (VEGF-C/D) and their receptors (VEGFR3) were detected both within and around tumors, and their contribution to lymph node metastasis is well documented. Indeed, expression of VEGF-C/D in cancer cells increases their metastatic capacity in animal models. Lymph nodes are also the major and early sites of metastasis a property of considerable prognostic significance in human cancers (eg, in breast, gastrointestinal, renal, colorectal, and other malignancies). These observations suggest that agents that block lymphangiogenesis may possess anticancer and antimetastatic activity (Tammela and Alitalo, 2010).
11.6.5 Tumor Initiating Cells and Blood Vessels
The ability to initiate clonal neoplastic growth, disease recurrence, and metastasis is often attributed to a subset of cancer cells known as CSCs, or TICs (see Chap. 13). There is an emerging relationship between CSCs and angiogenesis, of which two major scenarios are being considered. First, because the expression of stem cell properties and marker molecules (eg, Oct4) is, in some cases, induced by hypoxia, some CSCs may reside in poorly perfused tumor regions, possibly distant from the vasculature (Hill et al, 2009). In contrast, normal neuroectodermal stem cells and their malignant counterparts (CSCs) appear to be located in areas adjacent to blood vessels, and in regions referred to as perivascular niches (Gilbertson and Rich, 2007). In glioblastoma, brain TICs expressing CD133 antigen are found to exhibit elevated production of VEGF and an increased proangiogenic activity, largely as a result of the constitutive activation of the hypoxia response pathway mediated by HIF2 (Gilbertson and Rich, 2007). TICs may also exhibit altered procoagulant properties (Rak, 2009), and their abundance may be depleted following antiangiogenic therapies. Although the links between angio-genesis and CSCs are rather complex, their tumor initiating activity implicitly involves the vasculature (eg, during metastasis) (Gilbertson and Rich, 2007). CSCs have also been proposed to transdifferentiate to form tumor endothelium (Ricci-Vitiani et al, 2010), but this notion remains controversial.
11.7 BLOOD VESSEL-DIRECTED ANTICANCER THERAPIES
The concept of targeting blood vessels to achieve anticancer effects was initially developed by Judah Folkman (Folkman, 1971, 2007). After nearly 4 decades of experimental and clinical exploration, this modality is now a part of clinical management in several human cancers (see Table 11–2), with at least 6 different agents approved for human use by the Food and Drug Administration (FDA) in the United States. Targeting tumor blood vessels in cancer is based on functional and molecular differences that separate properties of the tumor microcirculation from the corresponding segments of the normal vasculature. Agents and techniques that target tumor blood vessels may act on ECs, or on mural cells directly (direct inhibitors). Alternatively, therapeutics may interfere with events and cells stimulating or supporting tumor vasculature (indirect inhibitors).
11.7.1 Antiangiogenic Therapies
Antiangiogenic agents are designed to block formation of new tumor blood vessels through several approaches ranging from targeting the specific signaling circuitry of ECs (eg, by blocking VEGF or VEGFR2), to exploiting naturally occurring angiogenesis inhibitory pathways (see Table 11–2). Whether a particular agent possesses an antiangiogenic activity can be determined using several functional angiogenesis assays conducted either on cultured ECs or in animals (Jain et al, 1997). Those tests analyze survival, proliferation, migration, and formation of tube-like structures by cultured ECs, ingrowth of capillaries into the chick chorioallantoic membrane, sprouting in avascular sites in animals (corneal micropocket assays), or in pellets of ECM (Matrigel) implanted subcutaneously into mice (Jain et al, 1997). In animal models and in the clinic antiangiogenic activity may also be deduced from tumor responses, as measured by functional imaging (see Chap. 14), for example, Doppler ultrasound, magnetic resonance (MRI), or positron emission tomography (PET), that allow the assessment of several relevant parameters, such as tissue metabolism, oxygenation, vascular permeability, or blood flow (Jain et al, 2009). Also changes in the density of blood vessels within the tumor mass (microvascular density [MVD] counts), shifts in circulating angiogenic cytokines (VEGF, IL-8), the presence of apoptotic ECs in peripheral blood, or changes in levels of CECs (EPCs) can be used in support of the antiangiogenic mechanism of a particular anticancer drug (Kerbel, 2008).
In spite of the growing understanding of biological events surrounding the antiangiogenic activity of various agents, the establishment of clinically useful predictive biomarkers for therapeutic antiangiogenesis has been challenging (Hanrahan et al, 2010). Nevertheless, hundreds of antiangiogenic agents have been evaluated thus far, and they can be broadly assigned into several main categories as summarized in Table 11–2 and Figure 11–10.
11.7.1.1 First-Generation, Exogenous Antiangiogenic Agents In early studies, several natural compounds or their derivatives were empirically found to possess activities against ECs. Examples of such agents include the fungal product fumagillin (and its derivative TNP470), penicillamine (copper chelator), carboxyaminotriazole, suramin, and 2-methoxyestradiol (2ME2) (Folkman and Kalluri, 2003). The mechanisms of action of many of these agents remain unclear. Thalidomide is an agent whose antiangiogenic activity is at least partially linked to the teratogenic effects of this drug and its grim earlier legacy (D’Amato et al, 1994). Thalidomide’s pleiotropic immunomodulating and antiangiogenic effects and those of the related agent, lenalidomide, are the basis of their recent use in the treatment of multiple myeloma and other malignancies (Cook and Figg, 2010).
11.7.1.2 Agents Based on the Activity of Endogenous Angiogenesis Inhibitors The ability of tissues to elaborate endogenous angiogenesis inhibitors inspired drug development aimed at therapeutic antiangiogenesis (Folkman, 2007). Several agents in this category have been evaluated, including antiangiogenic cytokines, such as interferon-α (IFNα), antiangiogenic fragments of ECM proteins (endostatin), fragments of plasminogen (angiostatin, K5), peptides related to TSP1 (ABT-510), and several other agents (Folkman and Kalluri, 2003). With few exceptions, the mechanisms of antiangiogenic activity of these agents are either complex, or have not been fully elucidated. In clinical settings, the therapeutic efficacy of these agents in cancer has been relatively modest, and only 1 drug in this category (YH-16-recombinant endostatin [Endostar]) is currently (2012) approved for human use, outside of North America (Cook and Figg, 2010).
11.7.1.3 Molecularly Targeted Antiangiogenic Agents Understanding of molecular pathways involved in angiogenesis has led to the development of several classes of targeted antiangiogenic agents. These can be broadly divided into 3 major categories: (a) neutralizing antibodies against angiogenic ligands, or their receptors, usually designated by generic names ending with “-mab” (eg, bevacizumab); (b) traps, which are agents designed to neutralize angiogenic factors by virtue of containing high affinity sites (ectodomains) derived from the corresponding cognate cellular receptors (eg, VEGF-trap/Aflibercept); and (c) small molecule inhibitors of angiogenic receptors, with generic names ending with “-nib” (eg, sunitinib).
Targeted antiangiogenic therapeutics in clinical use (and multiple experimental agents) are largely directed against the VEGF/VEGFR pathway. In 2004, the humanized monoclonal antibody directed against VEGF-A (bevacizumab) became the first antiangiogenic agent to be approved for treatment of metastatic colorectal cancer (Hurwitz et al, 2004). Subsequently, at least 5 additional small-molecule oral agents, targeting VEGF receptors and other RTKs (multikinase inhibitors), have entered medical practice in oncology (Table 11–2). Currently, VEGF/VEGFR antagonists are being used to treat several human cancers, even though limited long term activity and toxicity continue to present challenges. These key indications include metastatic renal cell carcinoma (MRCC), hepatocellular carcinoma (HCC), metastatic colorectal cancer (MCRC), non–small cell lung cancer (NSCLC), and glioblastoma (GBM). Antiangiogenics often provide a considerable, albeit mostly transient, benefit to at least a subset of patients (Kerbel, 2008). It is unclear why in other cancers (eg, in pancreatic adenocarcinoma or malignant melanoma) these agents are ineffective.
Additional angiogenic pathways, including PDGF/PDGFR (sunitinib, imatinib), FGF/FGFR (brivanib), Angiopoietin/TIE2 (AMG-386), HGF/Met (foretinib), EPHRINB2/EPH4 (JI-101), and DLL4/NOTCH (REGN421), are under investigation (see Table 11–2) (Cook and Figg, 2010). Targeted agents have also been developed against other molecular effectors of angiogenesis, with a more complex role, such as αvβ3/avβ5 Integrins (cilengitide), EGFL7 endothelial-binding ECM protein (MEGF0444A), and protein kinase (PK) C-β (enzastaurin) (Cook and Figg, 2010). Agents that are expected to have greater selectivity against cancer-related pathological angiogenesis and lymphangiogenesis, outside of common vascular pathways are also under development. Examples of such agents include inhibitors of PlGF (anti-PlGF antibody/TB-403), Bv8 (anti-Bv8 antibody), VEGF-C (VEGFR3-trap), VEGFR3 (anti-VEGFR3 antibody), PDGF-C (anti–PDGF-C antibody), and NRP1 and NRP2 (antibodies), as well as inhibitors of PHD2 (DMOG) and HIF (EZN-2968), the effects of which are not always restricted to ECs, and may involve mural, stromal and other cells (Carmeliet and Jain, 2011).
The exposure to antiangiogenic agents, especially those targeting VEGF, may trigger vascular remodeling. It is believed that downregulation, or blockade of VEGF acutely exposes ECs to the residual excess of ANG2. In the absence of VEGF-dependent survival/stimulating signals, ANG2 blocks the remaining TIE2 survival pathway and leads to endothelial cell apoptosis, resulting in vessel thrombosis, collapse, and regression (Hanahan, 1997). However, regression of capillaries does not obliterate the tissue “memory” of the prior capillary network. Instead, upon the disappearance of ECs tissue still contains the networks of their related basement membranes (capillary “sleeves”), which may serve as a scaffold for rapid endothelial regrowth upon restoration of VEGF supply, for example, once the antiangiogenic therapy has been discontinued (reviewed in Carmeliet and Jain, 2011).
11.7.2 Antiangiogenic Effects of Agents Targeting Oncogenic Pathways
The angiogenic switch in cancer cells represents another attractive therapeutic target. Examples are inhibitors of the protein farnesyltransferase, which block not only the transforming activity of mutant H-ras, but also VEGF upregulation driven by this oncogene in cultured cancer cells (Rak, 2009). Recent studies demonstrate similar effects of other oncogene-directed agents, including trastuzumab (HER2), cetuximab (EGFR), imatinib (BCR-ABL), and several others, an activity coupled with overt antiangiogenic mechanism demonstrable in vitro and in vivo (Rak, 2009). Moreover such agents may possess a dual activity against proangiogenic mechanisms operative in both cancer cells and in the endothelium. For example, farnesyltransferase inhibitors (tipifarnib, lonafarnib), or epidermal growth factor receptor (EGFR) inhibitors (gefitinib, erlotinib) may exert their effects by blocking farnesylation or EGFR signaling, respectively, in both cancer cell and endothelial compartments. Several anticancer agents under development inhibit 2 or more molecular targets (multikinase or “dirty” inhibitors). For example, by combining the inhibitory effects against VEGF and epidermal growth factor (EGF) receptors certain agents (vandetanib, AEE788) can act on both tumor cell and EC populations, including tumor-associated ECs that express abnormally high levels of EGFR. Indeed, combinatorial action of the multikinase inhibitors (eg, sunitinib or sorafenib) may explain some aspects of their clinical activity (Rak, 2009; Cook and Figg, 2010).
11.7.3 Antiangiogenic Effects of Other Therapeutics
The proangiogenic circuitry activated in endothelial, stromal, and cancer cells involves pleiotropic signaling pathways whose functions have often been studied outside of angiogenesis (see Chap. 8). Drugs that interfere with such pathways have been found to possess unsuspected antiangiogenic activities (Kerbel et al, 2000). Thus cyclooxygenase 2 (COX-2) regulates the expression of several angiogenic stimulators in cancer cells and the nonsteroidal antiinflammatory drugs (eg, celecoxib), which block this pathway, also possess antiangiogenic activity (Tsujii et al, 1998). Other pathways (agents) that are being studied in this context include mammalian target of rapamycin (mTOR; temsirolimus, everolimus), protea-some complexes (bortezomib), coagulation factors (heparin), steroid receptors, and antiangiogenic effects of anticancer chemotherapeutics.
The increased mitogenic activity of tumor-associated ECs may make them susceptible to traditional cytotoxic anticancer agents and radiation (Denekamp, 1982). These effects may be present in the context of the usual chemotherapy protocols, but they rapidly dissipate posttreatment because of vascular regrowth and mobilization of BMDCs. This occurs during “drug holidays” taken to allow for hemopoietic recovery (Kerbel, 2008). However, when chemotherapy drugs are given continuously at lower doses, their is reduced, drug holidays are no longer needed, and the antiangiogenic effects may be amplified, even in the presence of tumor cell resistance to the same agent. This approach is known as metronomic chemotherapy, and exhibits considerable antiangiogenic activity, especially when combined with VEGF inhibitors and other endothelial-targeting agents (Browder et al, 2000; Klement et al, 2000).
11.7.4 Vascular Disrupting Agents
While antiangiogenesis mainly targets new vascular growth, the already preformed tumor blood vessels are targets of another class of therapeutics, referred to as vascular disrupting agents (VDAs) (Tozer et al, 2005; McKeage and Baguley, 2010). The goal of this therapy is to provoke a selective vascular shutdown, or “infarction,” within the tumor vasculature, mainly by compromising the viability, continuity, and antithrombotic properties of the endothelial lining. Unlike antiangiogenesis, the effects of VDAs are rapid (begin within less than an hour), and result in acute intravascular thrombosis, vascular shutdown, ischemia, and tumor necrosis. Although these effects are dramatic in the center of the tumor mass, they dissipate in the periphery, leaving a viable rim of cancer cells supplied by the intact extratumoral vasculature, which is not susceptible to VDA treatment (Tozer et al, 2005). The residual disease and local vascular regrowth involving mobilization of cells from the bone marrow may lead to rapid tumor regrowth post-VDA treatment; this could be mitigated by combining VDAs with other agents, including inhibitors of angiogenesis and proper timing of these sequential treatments (Shaked et al, 2006).
The small-molecule VDAs are directed against vulnerable points of the tumor associated vasculature, especially tubulin and EC survival mechanisms (McKeage and Baguley, 2010). Two classes of these agents are under investigation (see Table 11–2), namely, (a) flavonoids (ASA404), which act mainly in a tubulin-independent manner, and (b) tubulin-binding agents. Some of the most studied compounds in this group include combretastatin A-4 phosphate (CA4P), AVE 8062, ABT-751, and OXi4503 (McKeage and Baguley, 2010).
11.7.5 Challenges Associated with Targeting Tumor Blood Vessels
11.7.5.1 Tumor-Specific Efficacy of Antiangiogenic Agents The distinct nature of tumor blood vessels, the quiescence of normal ECs, and their initially presumed absence of genetic instability were originally taken as an indication that, unlike conventional anticancer agents, therapies directed against blood vessels should be selective, nontoxic, universally applicable, and devoid of the risk of drug resistance. However, clinical experience with the approved agents indicated that their effects may vary considerably between different cancers and individual patients. Thus, inhibitors of the VEGF pathway (bevacizumab, sunitinib, sorafenib) have shown efficacy only in a narrow spectrum of specific diseases. Even if antiangiogenic therapy elicits initial response, relapse eventually occurs in most cases and in spite of continued treatment, a pattern reminiscent of the acquired resistance to other anticancer agents.
11.7.5.2 Mechanisms of Resistance to Blood Vessel-Directed Agents Acquired resistance to antiangiogenic agents can be a function of changes affecting ECs and their angiogenic mechanisms, or result from properties and abundance of host stromal cells. Such resistance can also be brought about by altered tumor microenvironment or genetic drift (or selection) of cancer cells. For example, progressive acquisition of oncogenic mutations may shift the expression of angiogenesis regulators leading to their increasing redundancy, and the failure of pathway-specific antiangiogenic agents (eg, VEGF inhibitors) (Rak, 2009). In addition, the expression of oncogenes and loss of tumor suppressors (eg, p53) may render cancer cells more capable of withstanding ischemic effects of antiangiogenesis (Yu et al, 2002). Examples of stromal cell-dependent resistance mechanisms that can circumvent VEGF inhibition include production of PDGF-C by fibroblasts, or Bv8 by tumor-associated granulocytes (Shojaei et al, 2009). Alternative mechanisms of angiogenesis (cooption, vasculogenic mimicry), increased vessel maturation (normalization), endothelial cell aneuploidy, autocrine reprogramming, rapid regrowth of blood vessels post-therapy, or influx of endothelial progenitors may all contribute to diminished therapeutic responses (Bergers and Hanahan, 2008). Hypoxic stress or production of cytokines may sometimes provoke more invasive behavior (or selection) amongst residual cancer cells, including increased metastasis. A better understanding of processes underlying resistance and invasiveness in this context is required to develop new more effective agents. Combinations between different antiangiogenic drugs and with other therapies may further improve the outcomes of this relatively new anticancer treatment modality.
11.7.5.3 Side Effects of Blood Vessel-Directed Agents Antiangiogenic agents are not absolutely selective and may block normal functions of angiogenic factors, or cause off-target effects that lead to toxicity. One common toxicity is the impairment of wound healing and the related risk of bleeding. In addition, VEGF pathway inhibitors may sometimes cause thrombosis, hypertension, and proteinuria as a consequence of their systemic impact on vascular homeostasis, vascular tone, or kidney function (Eremina et al, 2007). Other side effects may include fatigue, skin rash, hypothyroidism, and other poorly understood toxicities (Verheul and Pinedo, 2007).
11.7.5.4 Biomarkers of Therapeutic Response to Antiangiogenic Agents The chronic nature of therapeutic responses to antiangiogenic agents and their biological complexity make the effective monitoring of their anticancer effects challenging (Bertolini et al, 2010). Consequent therapeutic impact, individual responsiveness of a cancer patient, or early signs of drug resistance can sometimes be captured by using vascular imaging techniques and blood assays (Jain et al, 2009). The latter include measuring the levels of circulating endothelial-like cells, interleukins (eg, IL-6 or IL-8), TSP1, and other molecules (SDF1a, VEGF), and in some cases multiplex platforms that detect and profile several factors simultaneously (Jain et al, 2009). Still, new technologies are needed to improve the robustness and accuracy of antiangiogenic biomarkers for the expedited clinical development of new agents, and their more individualized use in cancer patients.
Related posts:
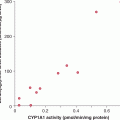
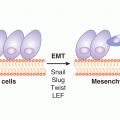
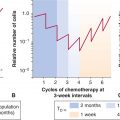
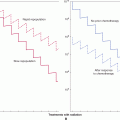
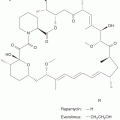
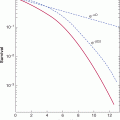
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


