Angiogenesis
Robert S. Kerbel
Lee M. Ellis
INTRODUCTION: ORIGINS OF THE CONCEPT OF ANTIANGIOGENIC THERAPY FOR CANCER
Among the most significant advances in medical oncology over the last 6 years is the U.S. Food and Drug Administration (FDA) approval of several antiangiogenic drugs for the systemic treatment of a variety of metastatic malignancies. Prior to the first successful randomized phase 3 clinical trial involving an antiangiogenic agent1 (bevacizumab, the humanized monoclonal antibody to vascular endothelial growth factor [VEGF]), enthusiasm in the field had waned by a combination of high expectations but little success in several pivotal phase 3 clinical trials. However, the initial success of bevacizumab, in combination with chemotherapy in the first-line treatment of metastatic colorectal cancer, initiated a resurgence in the field, in both the laboratory and the clinic, that has led to variable degrees of improvement in the therapy for advanced breast cancer, renal cell carcinoma (RCC), colorectal cancer, non-small cell lung cancer, glioblastoma, hepatocellular carcinoma, and ovarian cancer. However, despite these successes, clinical trials evaluating the use of these agents in some other malignancies has not yet led to similar success, and even where there is success, the clinical benefits are modest, as most notably in the case for breast cancer. Thus, it is essential to continue investigating basic mechanisms and mediators of angiogenesis with the aim of advancing the care of patients with malignant disease.
The era of antiangiogenic drug development began with the publication in 1971 of a landmark hypothesis article in the New England Journal of Medicine by M. Judah Folkman.2 He hypothesized that inhibition of blood vessel growth within a tumor could prolong tumor dormancy and improve survival of patients with minimal toxicity. Following publication of his hypothesis, Folkman and colleagues reported a significant number of discoveries that were instrumental in advancing the field, including defining the nature of the angiogenic “switch” in tumors, raising awareness of the presence of both pro- and antiangiogenic factors that mediate both pathologic and physiologic processes.3
Although the hypothesis of the essential role of angiogenesis in tumor growth, and the proposed therapeutic benefit of antiangiogenic drugs for cancer treatment, have been partially validated, there have been many surprising twists and turns in the field over the ensuing decades necessitating some interesting modifications of the basic therapeutic concept.4 These will be discussed in more detail later and include the following: (1) certain antiangiogenic drugs such as bevacizumab seem to have little or no clinical benefit when used as monotherapies to treat advanced disease of certain malignancies such as colorectal cancer, whereas they show clinical benefit only when used in combination treatments with other agents, particularly (thus far) chemotherapy4,5,6; (2) in addition to stimulators of angiogenesis, there are also a number of endogenous angiogenesis inhibitors, the expression of which may be down-regulated during tumor development, permitting a more robust angiogenic response3; (3) angiogenesis can also contribute to the growth of “liquid” hematologic malignancies, not just solid tumors3,5,7; (4) endothelial cells in developing blood vessels may be derived by incorporation and differentiation of systemically mobilized cells from the bone marrow, that is, circulating endothelial progenitor or precursor cells (CEPs) (Fig. 9.1.), not just by local division of pre-existing endothelial cells in resident vessels (“sprouting angiogenesis”)8,9; and (5) there is not a single “TAF” but a large and diverse array of molecular mediators of angiogenesis.
SEQUENTIAL STEPS INVOLVED IN THE FORMATION OF BLOOD VESSEL CAPILLARIES IN TUMORS
There are a number of sequential and fairly welldefined steps involved in the development for new capillary blood vessels, and their subsequent formation into a functional network. The first step in the formation of a capillary sprout (“sprouting angiogenesis”) from a pre-existing mature blood
vessel is the localized degradation of the surrounding basement membrane of the parental postcapillary venule. This creates a break to allow the movement of differentiated endothelial cells toward the adjacent tumor cells and the stimuli produced by such cells. Localized degradation is likely the consequence of the ability of various proangiogenic growth factors secreted by the tumor cell population or reactive stromal cells to induce synthesis and export of a number of proteolytic enzymes such as matrix metalloproteinases, cathepsins, and urokinase plasminogen activator.
vessel is the localized degradation of the surrounding basement membrane of the parental postcapillary venule. This creates a break to allow the movement of differentiated endothelial cells toward the adjacent tumor cells and the stimuli produced by such cells. Localized degradation is likely the consequence of the ability of various proangiogenic growth factors secreted by the tumor cell population or reactive stromal cells to induce synthesis and export of a number of proteolytic enzymes such as matrix metalloproteinases, cathepsins, and urokinase plasminogen activator.
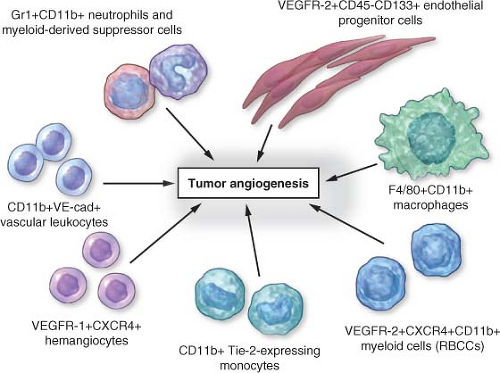 FIGURE 9.1 Circulating bone marrow-derived cell populations that stimulate or amplify tumor angiogenesis. The various hematopoietic (CD45-positive) cell types appear to have a perivascular location with respect to the tumor neovasculature, whereas the CD45-negative endothelial progenitor cells can become incorporated into the lumen of a growing blood vessel and differentiate into mature endothelial cells. In recent preclinical studies, neutrophils have also been shown to contribute to the induction of tumor angiogenesis. F4/80 is a pan macrophage cell-surface marker. CXCR4, CXC chemokine receptor 4; RBCCs, recruited bone marrow-derived circulating cells; VE-cad, vascular endothelial-cell cadherin (an adhesion molecule); VEGFR, vascular endothelial growth factor receptor. (From ref. 4, with permission.) |
The next step involves the directed locomotion/migration of endothelial cells from the parental venule toward the angiogenic stimulus emanating from the tumor mass. This is followed by division of endothelial cells that, in concert with migration, lengthen the “stalk” of the endothelial cell sprout. Subsequently, lumen formation takes place with completion of capillary sprouts and loops, and the envelopment of nascent capillaries with new basement membrane structures along with recruitment of perivascular support cells, especially pericytes. Critical in this process are specialized endothelial cells at the ends of growing capillaries called tip cells, which fuse with other tip cells to create a fused (linked) network of new capillaries.10 This sequence of events is thought to be quite similar to the formation of new blood vessel capillaries that occurs in developing embryos; however, the structure/morphology and function of many tumor-associated blood vessels can be highly irregular, heterogeneous, and functionally abnormal.6
Although this abbreviated description of sprouting angiogenesis is the most common view of angiogenesis, over the past 5 to 10 years, modifications or alternative views of angiogenesis have emerged. The mechanisms of angiogenesis may be organ- and/or tumor-specific. For example, in vascular-rich organs such as the brain, co-option may play an important role in providing a nutrient blood supply to the growing tumor.11 It has been hypothesized that parts of the vessel wall in tumors such as ocular melanoma or glioblastoma may be composed or melanoma cells, either in part (mosaic vessels)12 or full13 (“vascular mimicry”). Also noteworthy are the large number and diversity of molecular changes detected in
endothelial cells in tumor blood vessels during angiogenesis, many of which suggest possible new targets for development of antiangiogenic drugs.14,15 Some of these molecular changes might be related to the recently reported genetic and cytogenetic abnormalities detected in endothelial cells isolated from the tumor vasculature16 or by endothelial cell uptake of tumor cell-derived membrane vesicles.17
endothelial cells in tumor blood vessels during angiogenesis, many of which suggest possible new targets for development of antiangiogenic drugs.14,15 Some of these molecular changes might be related to the recently reported genetic and cytogenetic abnormalities detected in endothelial cells isolated from the tumor vasculature16 or by endothelial cell uptake of tumor cell-derived membrane vesicles.17
PERICYTES
Pericytes (our definition of pericytes is a single layer of periendothelial smooth muscle cells) modulate endothelial cell function, and are critical for the development of a mature vascular network. Pericytes regulate vascular function, including vessel diameter (and thus blood flow) and vascular permeability.18 Pericytes also provide mechanical support and stability to the vessel wall and maintain endothelial cell survival through direct cell-cell contact and paracrine circuits.18,19
The role of pericytes within the tumor vasculature is currently an intense area of study. The degree of pericyte coverage of endothelial cells in human tumors is controversial and discrepancies among studies may be because a single marker is not sufficient to examine pericyte presence and morphology. Markers such as alpha-smooth muscle actin, desmin, NG2, and RGS5 are commonly used, and confocal imaging is necessary to observe the true relationship of pericytes to endothelial cells.
Because of the role of pericytes in mediating endothelial cell survival,19 these cells have emerged as an important therapeutic target for antiangiogenic therapy. Studies of antiangiogenic agents targeting endothelial cell survival have demonstrated that such drugs result in increased apoptosis in endothelial cells that are not associated with pericytes, leading to a relative increase in the proportion of vessels with pericyte coverage.20 These data have led to the hypothesis that pericytes mediate resistance to antiangiogenic therapy. If this hypothesis is correct, targeting both endothelial cells and pericytes will increase the efficacy of antiangiogenic therapy, and there is some evidence in support of this hypothesis in preclinical studies.21,22 However, there is currently some growing doubt about the clinical impact of targeting pericytes using drugs such as multitargeting tyrosine kinase inhibitors (TKIs), for example, sunitinib, sorafenib, and pazopanib, which target platelet-derived growth factor (PDGF) receptors (which are expressed by pericytes) in addition to vascular endothelial growth factor receptors (VEGFRs). Thus, the therapeutic impact of such drugs does not appear to be significantly greater compared with specific anti-VEGF antibodies with the exception, currently, of hepatocellular carcinoma. Moreover, there is some limited preclinical evidence that inhibition of pericyte function and attachment to endothelial cells may actually facilitate metastasis by allowing tumor cell intravasation and extravasation.23 Still others have shown that inhibition of pericyte stimulatory factors may actually increase tumor growth, as pericytes may induce endothelial cell quiescence. Pericyte biology remains an important area of research that needs to be investigated more thoroughly.
DYSFUNCTIONAL NATURE OF THE TUMOR VASCULATURE
Although tumors possess the means to recruit and develop a new vascular network, this is not to suggest, as already mentioned, that such blood vessels are normal in either structure or function. Indeed, the characteristics of the vasculature in solid tumors are associated with a number of prominent abnormalities, the consequences of which have been hypothesized to have a significant impact on tumor growth, progression, and response to various anticancer therapies. For example, the structural and morphologic abnormalities include excessively dilated blood vessels, other vessels with areas containing absent or abnormal basement membranes, or having extreme corkscrewlike tortuosities, a relative lack of supporting perivascular cellular elements such as pericytes, or abnormalities in the pericyte population, and excessive vascular leakiness.24,25 These abnormalities can be quite variable within a solid tumor mass, and such heterogeneity can also extend to the relative density of blood vessels, which can be quite high in certain areas, and low in others.
As a result of all of these features, blood flow and perfusion within tumors can be highly heterogeneous and often sluggish, with some areas therefore being deprived of oxygen and nutrients leading to adjacent areas of elevated hypoxia. This may account for slow growth of tumors in some regions and more rapid growth in others. In addition, the marked leakiness/hyperpermeability of the tumor vasculature can lead to a marked extravasation of high-molecular-weight plasma proteins and fluid into the extracellular microenvironment within tumors, which can lead to elevated interstitial fluid pressures.24 It has been hypothesized that this can limit or retard the diffusion of anticancer drugs, especially antibodies or gene therapy vectors, and immune effector cells from the blood through the interstitium of the tumors.24 Thus, given their nature, tumor blood vessels, while necessary for progressive tumor growth and hematogenous metastasis, may also actually limit the efficacy of a broad and diverse array of anticancer drugs and treatments, including
chemotherapy, and oxygen required for optimal efficacy of radiation therapy.24
chemotherapy, and oxygen required for optimal efficacy of radiation therapy.24
MOLECULAR MEDIATORS OF TUMOR ANGIOGENESIS: ANGIOGENIC STIMULATORS AND THEIR RECEPTORS
Several diverse families of growth factors (angiogenic factors) are now known to stimulate/mediate tumor angiogenesis. Some, like VEGF,26 are primary, direct-acting factors that bind to cognate receptors that are primarily expressed on endothelial cells, especially when they are “activated.” Other factors are likely secondary in nature, that is, indirect acting. In other words, they stimulate expression of one or more of the primary proangiogenic growth factors or recruit cells to sites of angiogenesis that amplify the angiogenic process. Included in this group are such molecules as transforming growth factor beta (TGF-β), TGF-α, hepatocyte growth factor, inflammatory cytokines such as interleukin-6 and interleukin-8, cytokines such as granulocyte-colony stimulating factor, chemokines such as stromal-derived factor-1, and sex hormones such as estrogens and androgens.4 PDGFs have also been implicated as mediators of angiogenesis, for the most part through their effects on PDGF receptor-expressing pericytes, as previously mentioned. However, it is the primary, direct-acting factors, foremost among them VEGF,26 that are considered to be the principal driving forces in stimulating both physiologic and pathologic angiogenesis, including tumor angiogenesis, in most cases.
Direct-acting, primary proangiogenic growth factors include the VEGF family and their cognate receptor tyrosine kinases,26 the angiopoietins, especially angiopoietin-1 and -2 (ang-1/ang-2), and their cognate tyrosine kinase receptors, in particular tie-2 (see later discussion), and the Notch signaling receptor (specifically Notch 4) and its family of ligands, such as Deltalike ligand 4 (DLL4) and Jaggeds (see later discussion). All three systems have in common a high (but not absolute) degree of specificity for endothelial cells, and in particular activated endothelial cells associated with neovascularization. Another receptor tyrosine kinase-ligand system involved in angiogenesis is Eph receptor ephrin-B2.27 Ephrin-B2 is a transmembrane ligand that is involved in “bidirectional signaling” whereby Eph receptors help regulate endothelial tip cell guidance in the sprouting and branching of new blood vessel capillaries. Ephrin-B2 mediates its effects, at least in part, by regulating VEGFR-2 function.27
Discovery of the VEGF family and their receptors (Fig. 9.2) represented a profound turning point in the field of tumor angiogenesis research and the development of antiangiogenic drugs.26 Prior to the first published reports of VEGF, which was initially called vascular permeability factor,28 basic fibroblast growth factor (bFG)F was considered to be the central mediator of angiogenesis, and was the first molecular mediator of angiogenesis to be identified.3 However, bFGF lacks a signal sequence for cellular secretion, and therapeutic blockade of bFGF using antibodies did not cause consistent antitumor results, observations that raised doubts about a predominant role for bFGF in tumor angiogenesis.
VEGF was discovered in 1989 and reported to be a highly specific and potent mitogen for vascular endothelial cells.26,29 When the genes for vascular permeability factor and VEGF were sequenced, it was realized they were the same molecule.26 The vascular permeability function of VEGF is extremely potent (50,000-fold that of histamine) and probably accounts for much of the leakiness of the tumor vasculature. It is possible that enhanced permeability may be due to intercellular gaps between endothelial cells, decreased pericyte coverage (as a second barrier to permeability), and/or specialized endothelial cell organelles called vesiculovacuolar organelles,30 transmembrane vacuoles that can form channels leading to extravasation of fluid and proteins. VEGF (VEGF-A) is the prototypical member of a family of ligands with ˜40-80% homology: VEGF-A (also called simply VEGF), VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF) (Fig. 9.2). VEGF-A (hereafter called VEGF) actually exists in a number of variant isoforms based on RNA splicing. In humans, the most common splice variants are VEGF121, VEGF165, VEGF189, and VEGF206 (whereby the number denotes the number of amino acids in the mature protein). VEGF121, the shortest isoform, is freely circulating, whereas VEGF189 and VEGF206 are strongly bound to heparin sulphate containing glycoproteins and thus remain cell-bound or sequestered in the extracellular matrix where they remain biologically inactive until mobilized by specific proteases. VEGF165 has a heparin-binding sequence but can also freely circulate. Thus, VEGF121 and VEGF165 are generally considered to be the main VEGF family members that drive tumor angiogenesis. VEGF121 and VEGF165 bind to two tyrosine kinase receptors expressed by endothelial cells. These are known as VEGFR-1 (flt-1) and VEGFR-2 or KDR in humans (kinase insert domain receptor; with flk-1 being the KDR homolog in mice).26 The major signaling receptor is VEGFR-2. In contrast, VEGFR-1 signals only weakly, after VEGF binding, despite the fact that it can bind VEGF with tenfold greater affinity compared with VEGFR-2. A naturally occurring soluble form of VEGFR-1 is thought to serve as a negative regulator in physiologic angiogenesis. In addition, neuropilins (e.g., neuropilin-1 and neuropilin-2), which can bind class 3 semaphorins involved in axon guidance, can also bind the larger VEGF isoforms, as VEGF121 lacks the domain that binds to neuropilin.31 Neuropilin likely contributes to angiogenesis by serving as a coreceptor to VEGF and enhancing binding of VEGF-A to VEGFR-2.32 Antibodies that target both VEGF and neuropilin-1 yield better antiangiogenic responses than targeting a single protein.33
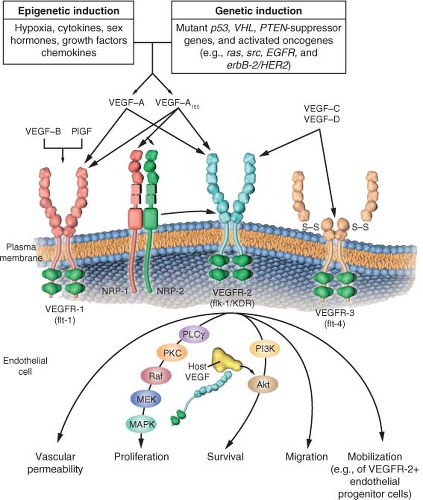 FIGURE 9.2 The family of vascular endothelial growth factor (VEGF) molecules and receptors. The major mediator of tumor angiogenesis is vascular endothelial growth factor A (VEGF-A, also called VEGF), specifically the circulating isoforms of VEGF, VEGF121 and VEGF165. These isoforms signal through VEGF receptor 2 (VEGFR-2), the major VEGF signaling receptor that mediates sprouting angiogenesis (called kinase-insert domain-containing receptor [KDR] in humans and fetal liver kinase 1 [flk-1] in mice). The role of VEGFR-1 in sprouting angiogenesis is much less clear. VEGF is expressed in most types of human cancer, and increased expression in tumors is often associated with a less favorable prognosis. Induction of or an increase in VEGF expression in tumors can be caused by numerous environmental (epigenetic) factors such as hypoxia, low pH, inflammatory cytokines (e.g., interleukin-6), growth factors (e.g., basic fibroblast growth factor), sex hormones (both androgens and estrogens), and chemokines (e.g., stromal cell-derived factor 1). Other causes include genetic inductive changes such as activation of numerous different oncogenes or loss or mutational inactivation of a variety of tumor suppressor genes. The binding of VEGF to VEGFR-2 leads to a cascade of different signaling pathways, two examples of which are shown, resulting in the up-regulation of genes involved in mediating the proliferation and migration of endothelial cells and promoting their survival and vascular permeability. For example, the binding of VEGF to VEGFR-2 leads to dimerization of the receptor, followed by intracellular activation of the phospholipase C gamma-protein kinase C-Raf kinase-MEK-mitogen-activated protein kinase (MAPK) pathway and subsequent initiation of DNA synthesis and cell growth, whereas activation of the phosphatidylinositol 3′-kinase (PI3K)-Akt pathway leads to increased endothelial cell survival. Activation of src can lead to actin cytoskeleton changes and induction of cell migration. VEGF receptors are located on the endothelial cell surface; however, intracellular (“intracrine”)-signaling VEGF receptors (VEGFR-2) may be present as well, and they are involved in promoting the survival of endothelial cells. The detailed structure of the intracellular VEGFR-2 in endothelial cells is not yet known, but it is shown as the full-length receptor that is normally bound to the cell surface. Binding of VEGF-C to VEGFR-3 mediates lymphangiogenesis. VEGF165 can bind to neuropilin (NRP) receptors, which can act as coreceptors with VEGFR-2 (horizontal arrow) to regulate angiogenesis. EGFR, epidermal growth factor receptor; flt-1, fms-like tyrosine kinase 1; PlGF, placental growth factor; PTEN, phosphatase and tensin homologue; S-S disulfide bond; VHL, von Hippel-Lindau. (From ref. 4, with permission.) |
Binding of VEGF to up-regulated endothelial cell VEGFR-2 sets in motion a unique intracellular signaling cascade.34 Various investigators have identified autophosphorylation on tyrosine residues in VEGFR-2, including residues 951, 1054, 1059, 1175, and 1214. Phosphorylation of Y1175 leads to activation of phospholipase C gamma, that in turn stimulates the protein kinase C (PKC) pathway leading to inositol trisphosphate generation and calcium mobilization. In addition, this pathway, via PKCβ, stimulates the c-Raf-MEK-MAP-kinase cascade.
Another member of the VEGF family, PlGF, binds to VEGFR-1, but not VEGFR-2, and there may be circumstances where it contributes to tumor angiogenesis. Interestingly, heterodimers of VEGF-A/PlGF may prevent angiogenesis by limiting VEGF-A signaling.35 VEGF appears to be a key mediator of embryonic angiogenesis, as well as both physiologic and pathologic forms of angiogenesis in the adult. A landmark discovery in this regard was the finding that disruption and inactivation of only one of the two VEGF alleles leads to embryonic lethality associated with marked developmental abnormalities of the vasculature (“haploininsufficiency”).36,37 Homozygous disruption of flk-1/VEGFR-2 or VEGFR-1 also leads to embryonic lethality accompanied by prominent vascular defects.26
There are at least four proposed roles by which VEGF is thought to promote tumor angiogenesis26: it can stimulate endothelial division, induce locomotion/migration, enhance endothelial cell survival38 by up-regulating various inhibitors of apoptosis,39 and mobilize endothelial progenitor cells from the bone marrow to sites of angiogenesis.4 In addition, the permeability enhancing effects of VEGF might also stimulate tumor angiogenesis by causing extravasation of large molecular proteins such as fibrinogen, that can be crosslinked to form a fibrin gel in the extracellular milieu of tumors serving as a matrix for endothelial cell migration and blood vessel formation.40 VEGF also has secondary effects including up-regulation of second messengers such as nitric oxide.
VEGF is expressed by most, if not all, human (and animal) cancers, often (but not always) at much higher levels than in corresponding normal tissues. Moreover, there are many reports showing elevated VEGF is an unfavorable prognostic marker.26 The ubiquitous and elevated expression of VEGF in both human and animal tumors is likely the consequence of many factors that are commonly associated with tumors, as shown in Figure 9.2. Among the most important is hypoxia, a prominent feature associated with the Gompertzian growth of solid tumors. Hypoxia can stabilize and hence up-regulate the levels of the hypoxia-inducible transcription factor called HIF1α, which in turn regulates hundreds of genes, among the most important of which is VEGF.41 In addition, a broad spectrum of oncogenes (e.g., ras, src, Her family members), and tumor suppressor genes, when they become mutated/inactivated or deleted, including p53, PTEN, and VHL, result in elevated VEGF expression.4
A second major growth factor signaling system that is known to be a major regulator of angiogenesis, especially for the later vessel maturation and stabilization stages, is the angiopoietin/tie-2 signaling pathway.42 There are a number of members in the family including angiopoietins-1-4 (Ang) with Ang-1 and -2 being the best characterized.43 Both of the latter bind to a highly specific endothelial cell-associated receptor tyrosine kinase, tie-2. Binding of Ang-1 to tie-2 causes an agonist effect whereas binding of Ang-2 is antagonistic. However, “pharmacologic levels” of Ang-2 may also serve as an agonist, which makes this system somewhat more complex and difficult to study.
Basic studies of this system suggest that Ang-1 is a stabilizing factor for endothelial cells; that is, it enhances endothelial cell survival and pericyte coverage. It is not truly a “proangiogenic” factor in the classic sense in that it does not promote endothelial cell proliferation. In fact, forced expression of Ang-1 in tumor cells leads to inhibited tumor growth from this “stabilizing” effect.44 In contrast, Ang-2 is a destabilizing factor that, if present with VEGF, can promote angiogenesis. Hence, Ang-2 is a rationale for inhibiting tumor angiogenesis.45 The tie-1 receptor remains an orphan receptor, with an undefined function. The Ang-1/tie-2 signaling pathway appears to be involved mainly in later stages of blood vessel formation, especially in the maturation and stabilization of vessels.46
Like VEGF and VEGF receptors, genetic disruption (“knockout”) of either Ang-1 or tie-2 leads to embryonic lethality, although both alleles of Ang-1 (or tie-2) have to be silenced (homozygous disruption), unlike VEGF.46 Studies of the role of Ang-1/tie-2 in tumor angiogenesis were hampered for many years by the inability to generate highly specific blocking antibodies or peptides. However, there are now a number of reports of the development not only of specific blocking peptides47 (“peptibodies”), which have proceeded to clinical trial assessment,45 but also monoclonal antibodies to Ang-2,48 which cause robust antitumor activity; such reagents should help considerably in clarifying the role of this system in tumor angiogenesis.
There are a number of other factors implicated in the process of angiogenesis including interleukin-8, epidermal growth factor receptor ligands, basic and acidic FGF, PDGF, among many others. However, because of the need for brevity in this chapter, we have focused on a number of factors most relevant to clinical medicine, and perhaps the future of oncology. Some of the aforementioned factors may take on increasing importance as mediators of resistance to drugs that target the VEGF pathway of angiogenesis.49,50
ENDOGENOUS INHIBITORS OF TUMOR ANGIOGENESIS
In addition to the existence of multiple molecular stimulators of angiogenesis, there are a large number of endogenous and intrinsic inhibitors of angiogenesis. The existence of such inhibitors was first surmised by Folkman3 on the basis of the observation that there are a number of tissues or organs that lack blood vessels and that also are rare sites of metastasis, such as cartilage or vitreous. It is also important to recognize the endogenous inhibitors are important in physiologic angiogenesis (wound healing, menstruation, luteal cycle) where a “stop” signal is necessary to prevent a pathologic condition.
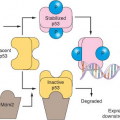
A breakthrough in the field of endogenous inhibitors came with a series of reports by Dameron et al.51 and Bouck et al.52 beginning in 1989/1990. They reported a large glycoprotein that is a prominent member of the extracellular matrix, namely, thrombospondin-1 (TSP-1), which binds to CD36 receptors, and is a potent endogenous angiogenesis inhibitor. Moreover, the p53 suppressor gene was found to up-regulate levels of TSP-1 in various cell types, and inactivation or loss of p53 is associated with down-regulation of TSP-1 expression,51 an observation that served to link the fields of cancer genetics—specifically the role of tumor suppressor genes and oncogenes in tumor development progression—with tumor angiogenesis.3,4 Subsequently, a number of other proteins were identified as endogenous inhibitors of angiogenesis.53,54,55,56,57,58 Many, if not most, are actually proteolytically cleaved fragments of larger proteins that are members of either the clotting/coagulation cascade family (e.g., angiostatin, which is a fragment of plasminogen53) or members of the extracellular matrix family of glycoproteins. Some examples of the latter category include endostatin,54 tumstatin, and canstatin, fragments of type IV collagen.55,56 Another endogenous inhibitor is known as vasostatin, which is a fragment of calreticulin.57 Vasohibin is a secreted protein that is produced by endothelial cells on stimulation with an angiogenic stimulator such as VEGF. Vasohibin was the first example of an endogenous inhibitor that operates on the principles of a negative feedback mechanism.58 A theory that has emerged from this body of work is that tumor angiogenesis likely requires two broad functional events: the induction or elevated expression of one or more proangiogenic growth factors, such as VEGF, coinciding with the down-regulation with one or more endogenous inhibitors, such as TSP-1.3,52,59
A COOPERATIVE REGULATOR OF TUMOR ANGIOGENESIS: THE NOTCH RECEPTOR-DLL4 SIGNALING PATHWAY IN ENDOTHELIAL CELLS
During the last 5 years, the Notch/Notch ligand system has been shown to mediate embryonic and tumor angiogenesis.60,61,62,63,64,65




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
