Alterations in Protein Metabolism in Diabetes Mellitus
Craig S. Stump
Sreekumaran Nair
HISTORICAL PERSPECTIVE
Before insulin treatment became readily available, stunted growth, cachexia, and muscle wasting were well recognized in patients with type 1 diabetes mellitus (1,2). Indeed, this association has been known for millennia, as documented in ancient Sanskrit literature describing diabetes mellitus as “honey-urine-disease” (3). The Greek physician Aretaeus described diabetes as a condition with “melting of flesh into urine.” Alternatively, the anticatabolic effect of insulin therapy has been well documented during the past 80 years (1,4,5). This anticatabolic effect is associated with normalization of urinary losses of nitrogen (4,6); increases in body cell mass, as indexed by total body potassium (7); and decreases in the concentrations of amino acids, especially of branched-chain amino acids (BCAA) (8,9). Intraarterial infusion of insulin during the postabsorptive state in subjects without diabetes decreases the net amino acid release from the forearm, indicating a net decrease in protein breakdown (10). Collectively, these observations demonstrate the important anabolic effects of insulin in humans. However, the mechanism by which insulin promotes the growth and maintenance of lean tissue continues to be debated, and the effects of insulin on the synthesis and breakdown of a variety of individual proteins remains an area of intense research.
This chapter reviews current knowledge of protein turnover in type 1 and type 2 diabetes mellitus. It describes the specific effects of insulin and other selected hormones on the components of protein turnover (i.e., protein breakdown and protein synthesis) and gives a general overview of the mechanisms by which insulin affects protein synthesis and breakdown within the cell. Although emphasizing human studies, it discusses important work from animal and in vitro studies and notes the discrepancies between different experimental approaches.
BALANCING PROTEIN SYNTHESIS AND BREAKDOWN
The balance between protein synthesis and breakdown determines the protein content of the whole body and individual tissues, as well as the concentrations of specific proteins. Consequently, protein turnover is not only important for increasing
and maintaining cell size but also fundamental to tissue remodeling, adaptation, and repair. Specific proteins are synthesized and catabolized at variable rates in response to particular functional demands upon the cell. In considering the proteins involved in the regulation of processes such as differentiation, growth, energy production, or gene repair, it is important to recognize that small alterations in protein balance may have large effects on cell function. Changes in the content, function, and integrity of specific proteins involved with these types of cellular processes probably contribute to the chronic complications associated with diabetes mellitus. However, an understanding of the underlying mechanisms is complicated by the regulation of protein turnover by a number of hormones other than insulin, including cortisol, glucagon, growth hormone, insulin-like growth factors and other growth factors, thyroid hormones, and testosterone. Substrates such as amino acids, fatty acids, and ketones and different physiologic states such as fasting, exercise, pregnancy, aging, and chronic disease also influence protein turnover. Recent advances in molecular biology and isotopic tracer technology have enhanced the capacity to study human protein metabolism. The human genome project provides information that sets the stage for a new era of investigation that not only will help define the functions of proteins encoded by newly identified genes but also will provide insight into their regulation and turnover dynamics. This undoubtedly will expand our understanding of the influence of diseases such as diabetes mellitus on protein metabolism and the impact of altered protein dynamics on the course of diabetes.
and maintaining cell size but also fundamental to tissue remodeling, adaptation, and repair. Specific proteins are synthesized and catabolized at variable rates in response to particular functional demands upon the cell. In considering the proteins involved in the regulation of processes such as differentiation, growth, energy production, or gene repair, it is important to recognize that small alterations in protein balance may have large effects on cell function. Changes in the content, function, and integrity of specific proteins involved with these types of cellular processes probably contribute to the chronic complications associated with diabetes mellitus. However, an understanding of the underlying mechanisms is complicated by the regulation of protein turnover by a number of hormones other than insulin, including cortisol, glucagon, growth hormone, insulin-like growth factors and other growth factors, thyroid hormones, and testosterone. Substrates such as amino acids, fatty acids, and ketones and different physiologic states such as fasting, exercise, pregnancy, aging, and chronic disease also influence protein turnover. Recent advances in molecular biology and isotopic tracer technology have enhanced the capacity to study human protein metabolism. The human genome project provides information that sets the stage for a new era of investigation that not only will help define the functions of proteins encoded by newly identified genes but also will provide insight into their regulation and turnover dynamics. This undoubtedly will expand our understanding of the influence of diseases such as diabetes mellitus on protein metabolism and the impact of altered protein dynamics on the course of diabetes.
EFFECTS OF INSULIN ON PROTEIN TURNOVER
Insulin has been shown to stimulate protein synthesis both in vitro and in situ (11,12,13,14,15,16). Studies in animals with low insulin levels, as occurs with fasting or β-cell destruction, have clearly shown that insulin replacement stimulates protein synthesis (12,17,18,19,20,21). Increased protein synthesis has been demonstrated in muscle tissue of nondiabetic animals after insulin administration in some (22,23) but not all (24) studies. The absence of an observed insulin effect in some reports may be related to the ages of animals studied and diminished insulin action in older animals (23,24,25). Studies on perfused or incubated rat diaphragm (26), skeletal muscle (12), and heart (17,21) have shown that insulin also decreases the breakdown of muscle protein.
The effects of insulin on protein metabolism in healthy human subjects without diabetes are less clear. In general, whole-body protein synthesis is not stimulated when insulin is infused during the postabsorptive state (fasting) (27,28,29). However, when insulin is infused under these conditions, circulating amino acids, especially BCAA, decrease in a dose-dependent manner (8,9,30). In contrast, when insulin is secreted in response to a meal under physiologic conditions, there is a simultaneous influx of dietary amino acids. Therefore, the lack of increase in protein synthesis observed in postabsorptive studies possibly relates to a decrease in the availability of amino acids. This concept is supported by studies that have examined the effects of infusing amino acids together with insulin in healthy subjects without diabetes (31,32,33,34,35,36). It generally has been observed that protein synthesis increases when insulin and amino acids are infused at rates sufficient to produce hyperinsulinemia and hyperaminoacidemia (32,36,37) but not with hyperinsulinemia and normal physiologic levels of amino acids (33). One study demonstrated that infusion of supraphysiologic doses of insulin together with amino acid supplementation can stimulate protein synthesis by more than 100% in forearm muscle of healthy subjects without diabetes (38). This protocol was designed to simulate, at the local muscle level, the high insulin concentrations used in many of the in vitro studies examining protein turnover. Therefore, the findings may help reconcile the observation of increased protein synthesis in vitro with the general absence of changes in protein synthesis in vivo in the presence of physiologic doses of insulin. In contrast to protein synthesis, the inhibitory effect of insulin on protein breakdown is readily demonstrated in subjects without diabetes even in the absence of infused amino acids (27,33,36,39,40). Similarly, several (27,28,39,41), but not all (42) studies measuring protein turnover in human limb muscle vascular beds have demonstrated decreased protein breakdown with insulin infusion. To summarize, findings in humans without diabetes concur with in vitro analyses and most animal studies in the demonstration of inhibitory effects of insulin on protein breakdown. By contrast, effects of insulin on protein synthesis in different experimental systems have not been consistent.
MECHANISMS OF INSULIN ACTION
Protein turnover in specific tissues and cell types is known to be affected differently in diabetes mellitus, depending on the degree of insulin deficiency, amino acid availability, and the level of glycemic control (40,43,44). It is likely that proteins in individual cellular subfractions (cytoplasm, mitochondria, nuclei, and contractile structures) respond differently to the effects of insulin and that these responses may be influenced by diabetes mellitus. The pathways by which insulin influences cellular processes such as gene transcription, messenger RNA (mRNA) stability, protein targeting for breakdown, and enzyme regulation undoubtedly are complex and exquisitely regulated. Knowledge of specific signaling pathways mediating insulin effects on protein synthesis and breakdown is advancing, and this new understanding will likely lead to practical new approaches to the management of the complications of diabetes mellitus.
Protein Synthesis
Although the role of insulin in regulating protein synthesis in humans remains uncertain, stimulation of protein synthesis by insulin in various in vitro models has been clearly shown (11,12,13,14,15,16). These somewhat divergent findings probably are related in part to methodologic issues. For example, it is difficult to show direct effects of insulin on gene expression and subsequent protein synthesis in intact animals because insulin deficiency and/or replacement results in concurrent changes in substrate concentrations and fluxes, changes in concentrations of counterregulatory hormones, and other factors. To circumvent this problem, investigators often attempt to maintain concentrations of glucose, amino acids, and various hormones by using clamp techniques. However, it is probably impossible to control all the known and still unknown factors that affect protein synthesis. For this reason, cell lines, primary cell cultures, tissue cultures, and in situ preparations represent useful systems for examining a more controlled cellular environment. These experimental models have the advantage of making it possible to evaluate insulin effects on gene expression in isolation or as a permissive or augmenting influence with other selected factors, but have the disadvantage of often uncertain physiologic relevance of the observed findings.
Insulin may influence protein synthesis through numerous sites of regulation (14,15), including effects on gene transcription (mRNA production), mRNA stability, ribosome biogenesis, initiation and elongation steps of mRNA translation, and the regulation of preexisting enzymes (Fig. 17.1). The effect of insulin
on gene transcription is usually viewed in terms of the cis / trans hypothesis, in which intracellular trans -acting factors bind to cis -acting DNA sites in the nucleus. The specific gene sites affected by insulin are referred to as insulin response sequences or elements (IRSs/IREs), which are usually located in the gene promoter region. Positive or negative effects can be mediated through these cis / trans interactions. Reputed IREs for a variety of genes, including glyceraldehyde 3-phosphate dehydrogenase, pyruvate kinase, glucagon, phosphoenolpyruvate carboxykinase (PEPCK), and insulin-like growth factor binding protein 1, have been identified (15). The transmission of the insulin signal to these genes could presumably upregulate the encoded protein, in isolation or in addition to the effects of insulin on general cellular protein synthesis, by producing more mRNA for translation. Examples of mRNAs that are selectively increased by insulin include eukaryotic elongation factor 2 (eEF-2) (45), phosphorylated heat- and acid-stable protein (PHAS-I) (46), and myosin heavy chain α (47).
on gene transcription is usually viewed in terms of the cis / trans hypothesis, in which intracellular trans -acting factors bind to cis -acting DNA sites in the nucleus. The specific gene sites affected by insulin are referred to as insulin response sequences or elements (IRSs/IREs), which are usually located in the gene promoter region. Positive or negative effects can be mediated through these cis / trans interactions. Reputed IREs for a variety of genes, including glyceraldehyde 3-phosphate dehydrogenase, pyruvate kinase, glucagon, phosphoenolpyruvate carboxykinase (PEPCK), and insulin-like growth factor binding protein 1, have been identified (15). The transmission of the insulin signal to these genes could presumably upregulate the encoded protein, in isolation or in addition to the effects of insulin on general cellular protein synthesis, by producing more mRNA for translation. Examples of mRNAs that are selectively increased by insulin include eukaryotic elongation factor 2 (eEF-2) (45), phosphorylated heat- and acid-stable protein (PHAS-I) (46), and myosin heavy chain α (47).
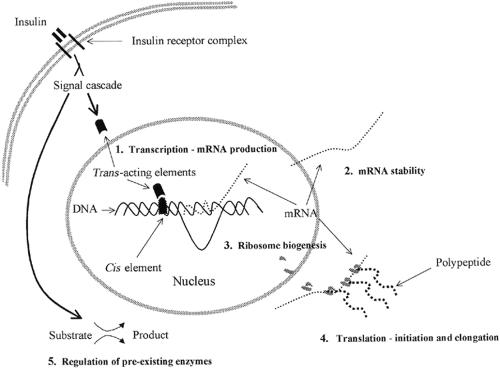 Figure 17.1. Sites of action of insulin in the regulation of protein synthesis. Insulin action depends on binding to the insulin receptor complex and initiation of a cascade of steps leading to its ultimate effect on protein synthesis. Insulin effects have been shown at various levels: (1) transcription of genetic message to mRNA; (2) contribution to mRNA stability; (3) biogenesis of ribosomes; and (4) translation—both at initiation and elongation. (See text for details of effects of each of these steps.) |
Insulin can also exert a selective influence on protein synthesis by stabilizing specific mRNA species. Degradation of mRNA can be measured by a decrease in radiolabel in an mRNA species prelabeled before the experiment, usually in the presence of transcription inhibitors. In these studies, stabilization of mRNA by insulin has been demonstrated for glycerol 3-phosphate dehydrogenase, glycogen phosphorylase, and glucose transporter 1 (GLUT1), among others (15,48). Conversely, insulin has been shown to destabilize some mRNAs, such as PEPCK (49,50) and GLUT4 (50). This leads to the interesting observation that insulin can stabilize some mRNA species and destabilize others within the same cell, further demonstrating the intricacy by which insulin regulates cell function. An example is the divergent effect of insulin on the stability of glycogen phosphorylase (48) and phosphoenolpyruvate carboxykinase mRNAs within hepatocytes (49). The specific mechanisms by which insulin influences mRNA stability are poorly understood at present.
Protein translation involves a complex set of reactions that decode the genetic information provided by mRNA into a polypeptide chain. Translation usually is divided into three phases: initiation, elongation, and termination. Protein-chain initiation involves binding of a methionyl-transfer RNA (tRNA) to a start codon in the mRNA, which in turn binds to 40S and 60S ribosomal subunits. The resulting mRNA-ribosome unit then adds tRNA-bound amino acid residues to the elongating peptide chain. Termination is the phase in which the completed polypeptide chain is released from the ribosome. Translational efficiency, particularly with regard to the initiation and elongation phases, has been a major focus of investigations of the effects of insulin on cellular protein synthesis. This is important because increases in translational efficiency have the potential to increase protein
synthesis without necessarily changing the total RNA content. The mechanisms by which insulin affects translation have been the topic of a recent comprehensive review (51). In general, this occurs through a series of interactions of the mRNA, ribosomal subunits, and specific binding and initiation factors, often involving the phosphorylation of one or more of these factors (52,53,54,55,56). Initiation starts with the binding of methionyl-tRNA to the 40S ribosomal subunit mediated by eIF-2 (51,57). It is noteworthy that eIF-2 will bind methionyl-tRNA only when the eIF-2 is complexed with guanosine triphosphate (GTP). Another initiation factor, eIF-2B, controls the recycling of eIF-2 between its GTP-bound and guanosine diphosphate (GDP)-bound forms. Methionyl-tRNA recruitment to the 40S ribosomal subunit is followed by mRNA binding and subsequent joining to the 60S ribosomal subunit. The resulting 80S initiation complex is then competent to enter the elongation phase. Insulin regulates the initiation of translation in part by enhancing the binding of mRNA to the 40S ribosomal subunit. One important step in this process requires attachment of the eIF-4E (cap-binding protein) and the eIF-4E-binding proteins PHAS-1 (4E-BP1) and eIF-4G to the 7-methyl guanine cap structure at the 5′ end of eukaryotic mRNAs (13,58,53,54). Insulin and perhaps other hormones stimulate this process by enhancing the phosphorylation of PHAS-1, dissociating it from eIF-4E so that eIF-4E can complex with eIF-4G, which associates with the 40S ribosomal subunit (58). Insulin has also been shown to increase the activity of eIF-2B, which regenerates eIF-2 as noted above (54,59). Rats made insulin deficient by blocking insulin secretion with diazoxide show a large decrease in the binding of eIF-4E to eIF-4G and an increase in eIF-4E * 4E-BP1 complex formation (56).
synthesis without necessarily changing the total RNA content. The mechanisms by which insulin affects translation have been the topic of a recent comprehensive review (51). In general, this occurs through a series of interactions of the mRNA, ribosomal subunits, and specific binding and initiation factors, often involving the phosphorylation of one or more of these factors (52,53,54,55,56). Initiation starts with the binding of methionyl-tRNA to the 40S ribosomal subunit mediated by eIF-2 (51,57). It is noteworthy that eIF-2 will bind methionyl-tRNA only when the eIF-2 is complexed with guanosine triphosphate (GTP). Another initiation factor, eIF-2B, controls the recycling of eIF-2 between its GTP-bound and guanosine diphosphate (GDP)-bound forms. Methionyl-tRNA recruitment to the 40S ribosomal subunit is followed by mRNA binding and subsequent joining to the 60S ribosomal subunit. The resulting 80S initiation complex is then competent to enter the elongation phase. Insulin regulates the initiation of translation in part by enhancing the binding of mRNA to the 40S ribosomal subunit. One important step in this process requires attachment of the eIF-4E (cap-binding protein) and the eIF-4E-binding proteins PHAS-1 (4E-BP1) and eIF-4G to the 7-methyl guanine cap structure at the 5′ end of eukaryotic mRNAs (13,58,53,54). Insulin and perhaps other hormones stimulate this process by enhancing the phosphorylation of PHAS-1, dissociating it from eIF-4E so that eIF-4E can complex with eIF-4G, which associates with the 40S ribosomal subunit (58). Insulin has also been shown to increase the activity of eIF-2B, which regenerates eIF-2 as noted above (54,59). Rats made insulin deficient by blocking insulin secretion with diazoxide show a large decrease in the binding of eIF-4E to eIF-4G and an increase in eIF-4E * 4E-BP1 complex formation (56).
Insulin may increase protein synthesis by selectively upregulating the initiation of translation of mRNAs containing a polypyrimidine tract at their 5′ transcriptional start site. These mRNAs encode important components of the translational apparatus, including ribosomal proteins and translational elongation factors that would likely increase the overall capacity of the cell for protein synthesis. Translation of such 5′-polypyrimidine mRNAs appears to be regulated in part via multisite phosphorylation of the 40S ribosomal protein S6 by p70 S6 kinase (p70S6K) (60,61). An increase in S6 phosphorylation has been observed in a variety of cells in culture exposed to insulin [reviewed in references (14,62,63,64)], and S6 phosphorylation generally correlates with increased protein synthesis (63). Investigators have shown that expression of a dominant interfering mutant of p70S6K, which prevents the activation of wild-type p70S6K, blocks serum-activated translation of 5′-polypyrimidine tract mRNA in human kidney 293 cells (61).
The elongation phase of protein synthesis involves the progressive addition of amino acyl-tRNAs to the ribosomal mRNA complex, causing a lengthening of the peptide chain until termination. At least two elongation factors (eEF-1 and eEF-2) are important in facilitating this process. The eEF-1 factor is complexed with GTP in a fashion similar to that described above for eIF-2, and GTP-bound eEF-2 mediates the attachment of amino acyl-tRNA to the ribosome. Subsequently, eEF-2 is required for the movement of the ribosome relative to the mRNA and for migration of the amino acyl-tRNA from the acceptor site to the peptidyl site of the ribosome (65). Like eEF-1, eEF-2 is a phosphoprotein and is regulated by insulin. Phosphorylation of eEF-2 by way of eEF-2 kinase results in its complete inactivation (66). Insulin appears to increase the rate of elongation by the inactivation of eEF-2 kinase through a pathway sensitive to the antifungal agent rapamycin, which is discussed in greater detail below (67). Collectively, these studies indicate that insulin may enhance important steps in translation initiation and elongation. The significance of these steps in insulin-stimulated protein synthesis and the steps that are rate limiting remain to be determined.
The capacity of the cell to synthesize protein can be determined in part by the abundance of ribosomes. As noted above, ribosomes are essential components of the translational machinery, and their biogenesis involves the synthesis of approximately 80 ribosomal proteins and four rRNA species transcribed by RNA polymerases I (18S, 28S, 5.8S) or III (5S) (68,69). Production of ribosomal precursor molecules is dependent on the stage of cellular development, nutrient levels, growth factors, and hormones (69,70). Nucleoli contain the rRNA genes and are the sites of rRNA synthesis, processing, and assembly into ribosomes. To accommodate the demand for rapid production of large numbers of ribosomes, the rRNA transcription units are repeated in tandem up to 1,000 times in eukaryotic nucleolar sites (71). From the nucleolus, the components must be packaged and moved to the cytoplasm, where translation occurs. Insulin has been shown to increase the synthesis of ribosomal proteins fourfold in chick embryo fibro-blasts (72) and to stimulate ribosomal protein synthesis in mouse myoblasts (73).
Although the mechanisms by which insulin increases the cellular content of functional ribosomal complexes are still largely unknown, there is evidence for a significant effect of insulin on posttranscriptional events (72,73). For example, the addition of insulin results in a selective increase in the fraction of ribosomal protein mRNAs associated with polysomes in mouse myoblasts (73), and the association of polysomes with the cellular cytoskeleton is increased in response to insulin when protein synthesis increases (74,75). The synthesis of rRNA species also is stimulated by insulin in a variety of cell types, including fibroblasts (72,76), myoblasts (73), and hepatocytes (77). Changes in rDNA transcription can involve changes in chromatin structure to permit the formation of initiation complex, alterations in RNA polymerase activity, or alterations in associated transcription factors. Insulin-stimulated increases in rRNA transcription in mouse fibroblasts and a rat hepatoma cell line are not associated with a change in the cellular content of RNA polymerase I (76). However, increases in upstream binding factor (UBF) and RNA polymerase I-associated factor 53 have been observed; both appear to aid in recruitment of RNA polymerase I to the rDNA promoter. It is interesting that overexpression of UBF in mouse fibroblasts was able to directly stimulate rDNA transcription (76). Finally, insulin may also diminish the rate of ribosome degradation (14,77,78).
The transmission of the insulin signal from outside the cell through the plasma membrane and cytoplasm and into the nucleus, where it can influence trans -acting factors, is an important component of insulin action. At least two general pathways by which insulin may affect gene transcription have been suggested. The first is the classical signal transduction in which formation of an insulin-receptor complex at the plasma membrane transmits a cascading signal through the cytoplasm into the nucleus via second-messenger molecules and protein phos-phorylation/dephosphorylation reactions. The second, and less firmly established, pathway involves the effects of insulin on intracellular receptors (79). This hypothesis is supported by several reports of mediation of changes in transcription by internalized insulin (79,80,81) through nuclear insulin receptors (82,83). Whether direct insulin interactions with nuclear receptors and DNA IREs have important effects on protein synthesis is unknown.
Although direct nuclear actions may be significant, insulin is thought to exert most of its effects by changing the phosphorylation state of various regulatory proteins important to protein synthetic pathways via classical signal transduction (14). This
pathway involves autophosphorylation of insulin receptors initiated by the binding of insulin, followed by increased tyrosine phosphorylation of intracellular proteins such as insulin-receptor substrates (IRS-1, IRS-2, IRS-3, and IRS-4) and the protein Shc. IRS-1 is the best characterized of the IRS family of proteins. Its phosphorylated tyrosine sites are thought to associate with high-affinity src homology 2 domain (SH2)-containing proteins such as on the 85-kDa regulatory subunit (p85) of phosphoinositide 3-kinase (PI 3-kinase). Of note, both the insulin receptor and IRS-1 appear to be required for the insulin-mediated stimulation of general and growth-related protein synthesis (84). PI 3-kinase is emerging as the only phosphotyrosine-activated effector downstream of IRS-1 that is essential for protein synthesis (84,85,86,87). LY 294002, a specific inhibitor of PI 3-kinase, completely prevents insulin-mediated stimulation of protein synthesis in rat skeletal muscle (86). A number of other effectors downstream of IRS-1 are also being investigated for their roles in insulin signaling to protein synthesis, including protein kinase C (59,88), ras (89), mitogen-activated protein (MAP) kinases (85), p70S6K (85), and PHAS-1 (13,55,86,90).
pathway involves autophosphorylation of insulin receptors initiated by the binding of insulin, followed by increased tyrosine phosphorylation of intracellular proteins such as insulin-receptor substrates (IRS-1, IRS-2, IRS-3, and IRS-4) and the protein Shc. IRS-1 is the best characterized of the IRS family of proteins. Its phosphorylated tyrosine sites are thought to associate with high-affinity src homology 2 domain (SH2)-containing proteins such as on the 85-kDa regulatory subunit (p85) of phosphoinositide 3-kinase (PI 3-kinase). Of note, both the insulin receptor and IRS-1 appear to be required for the insulin-mediated stimulation of general and growth-related protein synthesis (84). PI 3-kinase is emerging as the only phosphotyrosine-activated effector downstream of IRS-1 that is essential for protein synthesis (84,85,86,87). LY 294002, a specific inhibitor of PI 3-kinase, completely prevents insulin-mediated stimulation of protein synthesis in rat skeletal muscle (86). A number of other effectors downstream of IRS-1 are also being investigated for their roles in insulin signaling to protein synthesis, including protein kinase C (59,88), ras (89), mitogen-activated protein (MAP) kinases (85), p70S6K (85), and PHAS-1 (13,55,86,90).
There appear to be essentially two mechanisms of insulin-stimulated protein synthesis, one controlled by PI 3-kinase and the other by the GTP-binding protein p21ras (87,88). The insulin-stimulated PI 3-kinase pathway appears to exert its effect primarily by way of the Ser/Thr protein kinase Akt and subsequent activation of p70S6K(91) and PHAS-1 (92). Akt and p70S6K can be activated directly by the products of PI 3-kinase, phosphatidyl-inositol 3,4-bisphosphate and phosphatidylinositol 3,4,5-tris-phosphate (93). The use of the antifungal agent rapamycin helps in characterizing the pathway downstream of PI 3-kinase and Akt by acting at the level of TOR (target of rapamycin; mTOR in mammalian cells), a novel protein kinase, which appears to be on the pathway between Akt and both p70S6K and PHAS-1. Rapamycin inhibits activation of both of these factors (46,84,86,88,94) and attenuates insulin-stimulated protein synthesis (13,85,86). Alternatively, there appears to be a rapamycin-independent pathway involving an atypical protein kinase C isoform (PKCζ) downstream from PI 3-kinase involved in insulin-stimulated protein synthesis (88). It has been suggested that one pathway, through PKCζ, leads to general or “housekeeping” protein synthesis, whereas the other pathway, through Akt, stimulates growth-regulated protein synthesis (88).
Insulin stimulates Ras activity by two known mechanisms that involve binding the Grb/SOS complex by either Shc or IRS-1 (51,95) and subsequently leading to the formation of the active GTP-bound state of Ras. Ras then stimulates a cascade of protein serine/threonine kinase events that involve Raf, MEK, and the MAP kinases ERK1 and ERK2 (96,97). However, it is uncertain whether the activation of the MAP kinases contributes significantly to insulin-stimulated protein synthesis (13,84,98). Furthermore, it is important to note that the PI 3-kinase and p21rasbranches are not completely separate and that there is evidence that PI 3-kinase may be upstream of ras under some conditions (87,99,100). Of interest is the finding that increases in IRS-1 tyrosine phosphorylation (101), PI 3-kinase activity (102), p85–PI 3-kinase mRNA expression (101), and Akt kinase activity (103) in response to insulin are diminished in skeletal muscle of patients with type 2 diabetes.
Protein Breakdown
A normal 70-kg adult synthesizes and degrades approximately 280 g of protein each day under standardized conditions (104). Most of this protein turnover occurs within cells. Intracellular protein turnover is continuous, albeit at vastly different rates (i.e., different protein half-lives), depending on the identity, function, and location of the protein, as well as on the physiologic state of the organism. Catabolic states such as acute starvation or untreated type 1 diabetes result in increased protein breakdown, particularly from skeletal muscle, which is an important reservoir of amino acids under these conditions. The resulting increase in circulating amino acids provides precursors for protein resynthesis and for gluconeogenesis in the liver, and the BCAA offer an additional energy source for the remaining muscle tissue. The amino acids from protein breakdown also are crucial for maintaining synthesis rates of essential proteins such as clotting factors produced by the liver. Fortunately, the regulation of cellular protein breakdown is highly specific and precise—often able to select less essential proteins for breakdown, to the overall benefit of the organism. The regulatory mechanisms responsible for this specificity in protein breakdown remain to be defined. Rapid or sustained breakdown of protein, as occurs in starvation, catabolic disease states, and other severe physiologic stresses, may result not only in losses in muscle mass but also in an overall disruption of homeostasis. Understanding the cellular mechanisms involved in protein breakdown is an essential step toward developing pharmacologic or other interventions that might reduce excessive losses of essential proteins during catabolic conditions.
It is important to emphasize that the influence of protein breakdown extends to the regulation of cell function under all physiologic conditions and not just to catabolic states. Generally, this essential regulatory process is observed as enzymes, trans-cription factors, receptors, and other signaling proteins undergo selective degradation. For example, the levels of gluconeogenic enzymes, such as PEPCK, increase in rat liver with fasting and promptly decrease after refeeding (105,106). Such rapid fluctuations in hepatic cell enzyme content require a significant change in the ratio of protein breakdown to protein synthesis. Another example of the mediation of metabolic change by protein breakdown is evident in the degradation of enzymes such as glutamine synthetase, ornithine decarboxylase, and 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase in response to the accumulation of their reaction products (107). Alternatively, a decrease in the breakdown of a family of proteins known as heat-shock proteins (e.g., hsp70) occurs when cells are exposed to heat treatment (108). Heat-shock proteins appear to be cytoprotective during a variety of cellular insults, in part by facilitating the recovery of metabolic pathways that have been perturbed by cellular stress (109). It is interesting that hsp70 and cofactors known as DnaJ homologues are themselves essential in the preparation of certain abnormal proteins for breakdown in eukaryotic cells (110). In summary, protein breakdown is essential for cellular well-being through its roles in eliminating potentially lethal accumulations of abnormal, damaged, or foreign proteins; promoting normal tissue remodeling and repair; facilitating regulation of numerous metabolic pathways; and providing nutritive substrates in catabolic states. Defects in protein breakdown mechanisms clearly have the potential to cause abnormal tissue growth, whereas accelerated protein breakdown can result in tissue or organ wasting.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


