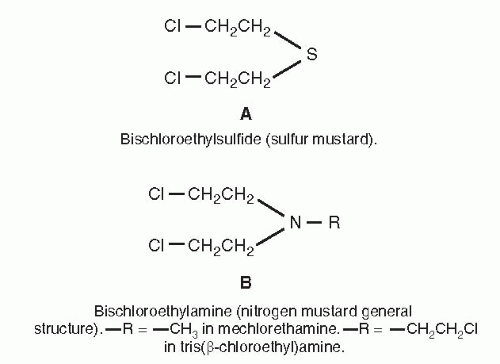
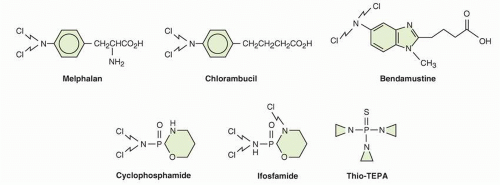
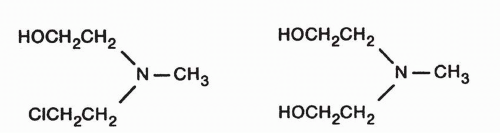
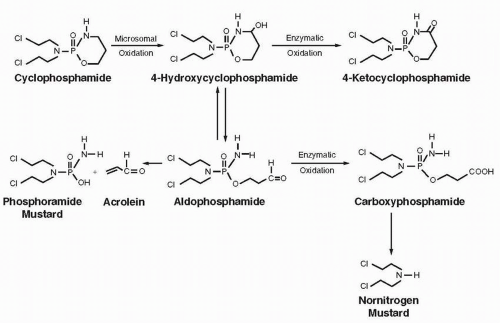
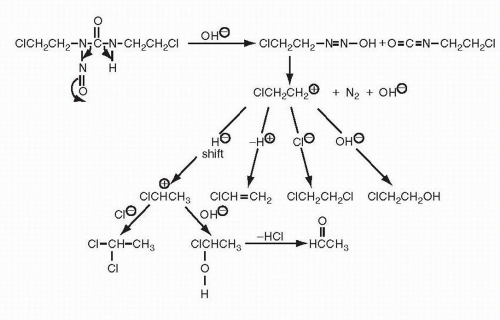
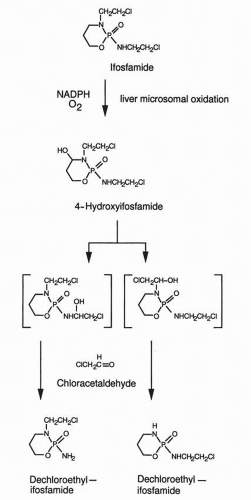
Cellular Uptake
The uptake of alkylating agents into cells is an important determinant of cellular specificity. Many are highly lipid soluble (including the active metabolites of the methylating agents, cyclophosphamide, and ifosfamide, as well as chlorambucil) and readily enter cells by passive diffusion. Mechlorethamine uptake depends upon the choline transport system.
56 Melphalan is transported into several cell types by at least two active transport systems, which also carry leucine and other neutral amino acids across the cell membrane.
57,
58 High levels of leucine in the medium protect cells from the cytotoxic effects of melphalan by competing with melphalan for transport.
59 In contrast to mechlorethamine and melphalan, the highly lipid-soluble nitrosoureas BCNU and CCNU enter cells by passive diffusion.
60 Chlorambucil uptake also occurs through simple passive diffusion.
Studies of cellular uptake of alkylating agents that require metabolic activation (such as cyclophosphamide or ifosfamide) are hampered by uncertainty about which metabolite, or even parent drug, is the most critical moiety for transport.
Sites of Alkylation
Any alkylating agent producing reactive intermediates binds to a variety of cellular constituents
61 including nucleic acids, proteins, amino acids, and nucleotides. As an example, the active alkylating
species from a nitrogen mustard demonstrates selectivity for nucleophiles in the following order: (a) oxygens of phosphates, (b) oxygens of bases, (c) amino groups of purines, (d) amino groups of proteins, (e) sulfur atoms of methionine, and (f) thiol groups of cysteinyl residues of glutathione.
62 This ranking, however, assumes there are no steric or hydrophilic/hydrophobic barriers to the tissue nucleophile, and this is seldom the case. In addition, glutathione conjugation is often favored in the presence of GSTs, which offer catalysis. Thus, generalizations about alkylating agent targets are fraught with difficulty. In addition, it seems likely that a matrix of biochemical targets of alkylating agents may contribute to cytotoxicity, though DNA is generally favored as the primary target. Proof of this hypothesis may be emerging from three areas of research where cytotoxicity correlates with (a) activity of DNA repair enzymes, perhaps best shown for BCNU and repair by alkyl guanine alkyltransferase (AGT),
63 (b) changes in a matrix of genetic and epigenetic events measured and analyzed by gene expression arrays,
64 and (c) specific DNA adducts shown by mass spectrometric analysis.
65 The stringency of such analyses requires that alternative toxic pathways not involving DNA must be excluded, a difficult requirement to meet. For this reason, mechanistic understanding of alkylating agent activity must be considered incomplete.
In the DNA molecule, the phosphoryl oxygens of the sugar phosphate backbone are obvious electron-rich targets for alkylation. Alkylation of the phosphate groups occurs
66,
67 and can result in strand breakage from hydrolysis of the resulting phosphotriesters. Although the biologic significance of the strand breakage caused by phosphate alkylation remains uncertain, the process is so slow that it seems unlikely to be a major determinant of cytotoxicity, even for monofunctional agents.
68Extensive studies with carcinogenic alkylating agents such as methyl methane sulfonate have shown that virtually all the oxygen and nitrogen atoms of the purine and pyrimidine bases of DNA can be alkylated to varying degrees. The relative significance of these sites of alkylation of specific bases and of specific sites on DNA in determining cytotoxicity, specific organ toxicities, or carcinogenesis remains uncertain. Alkylation of the O-6 atom and of the extracyclic nitrogen of guanine appears to be particularly important for carcinogenesis.
69,
70,
71Studies of the base specificity of alkylation by the chemotherapeutic alkylating agents have been much less extensive. Busulfan and mechlorethamine alkylate the N-7 position of guanine. Guanine cross-links (two guanine molecules abridged at the N-7 position by an alkylating agent) have been isolated from acid hydrolysates of the reaction mixtures.
35,
36Reaction of the nitrogen mustard with native DNA, however, produces alkylation of the N-1 position of adenine in addition to N-7-alkylated guanine. The enhanced alkylation of the N-7 position of guanine may result from base stacking and charge transfer that enhance the nucleophilic character of the N-7 position.
72 Melphalan preferentially alkylates guanine N-7 or adenine N-3.
65Base sequence influences the alkylating reaction. The N-7 position of guanine is most electronegative and, therefore, most vulnerable to attack by the aziridinium cation intermediate of the nitrogen mustards when the base is flanked by guanines on its 3′ and 5′ sides. The key site of DNA attack for the nitrosoureas as well as nonclassic methylating agents such as procarbazine and dacarbazine seems to be the O-6 methyl group of guanine.
7 Enhanced repair of this site is associated with drug resistance.
73 Thus, the preferred sites for alkylation vary by alkylating agent and chemical environment around the DNA base in question.
DNA Cross-Linking
On the basis of their isolation of the guanines linked at N-7 by alkylating agents, Brookes and Lawley
72,
74 postulated that the bifunctional alkylating agents such as the nitrogen mustards produced interstrand and intrastrand DNA-DNA cross-links and that these cross-links were responsible for the inactivation of the DNA and cytotoxicity. On the basis of the Watson-Crick DNA model, these authors suggested that appropriate spatial relationships for cross-linking by nitrogen mustards or sulfur mustard occurred between the N-7 positions of guanine residues in complementary DNA strands (
Fig. 14A-7).
The importance of cross-linking is supported by the fact that the bifunctional alkylating agents, with few exceptions, are much more effective antitumor agents than the analogous monofunctional agents. Furthermore, increasing the number of alkylating units on the molecule beyond two does not usually increase the antitumor activity of the compound.
Direct evidence that DNA cross-linking occurs as the result of treatment of DNA or cells with bifunctional alkylating agents was provided initially by relatively insensitive physical techniques, including sedimentation velocity studies and denaturation-renaturation studies.
75 These techniques, however, could not detect DNA interstrand cross-linking in mammalian cells exposed to therapeutic levels of alkylating agents in vitro or in tissues after in vivo drug administration. In 1976, a more sensitive assay for DNA interstrand cross-linking in cells, the alkaline elution method,
76 was reported and had the necessary sensitivity to detect DNA cross-linking in cells and tumor-bearing animals exposed to minimal cytotoxic levels of alkylating agents.
77,
78 These studies and others using ethidium bromide fluorescence to detect cross-links have shown that DNA cross-linking by bifunctional alkylating agents correlates with cytotoxicity and that DNA in drug-resistant cells has lower levels of cross-linkage.
79,
80 The alkaline elution technique also detects DNA-protein as well as DNA-DNA cross-links.
81 DNA-protein crosslinks likely do not play a major role in cytotoxicity and may be repaired by replication bypass mechanisms.
81In addition to these target effect-response studies, silencing of the AGT promoter in gliomas correlates with improved antitumor activity and survival in patients treated with BCNU.
82 Because AGT repairs guanine alkylation products produced by BCNU, decreased enzyme activity would be expected to increase DNA alkylation, implying that DNA is a critical target for BCNU effects. Thus, evidence increasingly supports the hypothesis that DNA adduct formation is the major mechanism of alkylating agent cytotoxicity.
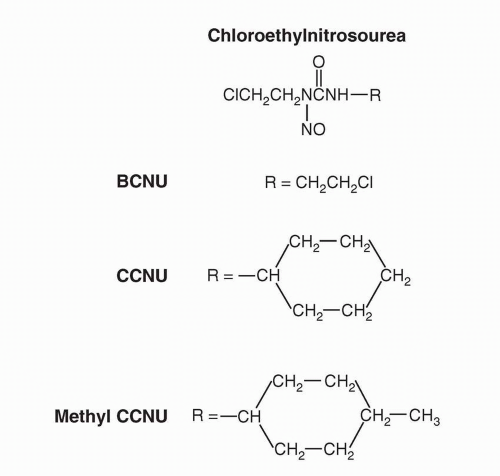
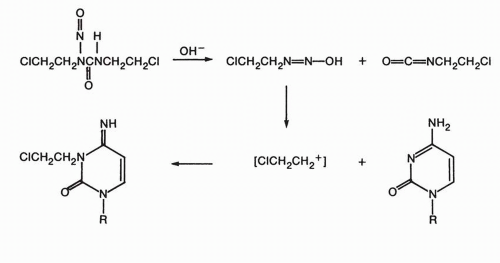
Chloroethylnitrosoureas cross-link via a unique mechanism.
43 The spontaneous decomposition of the chloroethylnitrosoureas generates a chloroethyldiazonium hydroxide entity that can alkylate DNA bases to produce an alkylating chloroethylamine group on the nucleotide in the DNA strand. This group could then alkylate an adjacent nucleotide on the complementary DNA strand in a slower step, producing an interstrand cross-link. The mechanism of alkylation by thiophosphates such as thiotepa likely begins with protonation of the aziridine N, which leads to ring opening. Cross-linking can proceed by one of several mechanisms, either activation of the free chloroethyl carbon or activation of a second aziridine ring on the original molecule. Although interstrand cross-links are important mediators of the cytotoxic effects of alkylating agents, the monofunctional DNA alkylations exceed cross-links in number and are potentially cytotoxic. This hypothesis is supported by the fact that certain clinically effective agents, such as procarbazine and dacarbazine, are monofunctional alkylating compounds and do not produce cross-links in experimental systems. The basis of the cytotoxic effects of monofunctional alkylation appears to be through mismatch repair-mediated processes, since cells lacking mismatch repair are often methylating agent tolerant. The futile cycling of repair reactions through mismatch repair efficiently removes bases opposite
O6-methylguanine and reinserts a thymine, with repeated attempt to repair the
O6-mG:T mismatch and further futile repair. This produces single and double strand breaks that are cytotoxic, especially in the second round of DNA synthesis, after formation of the
O6-mG:T mismatch.
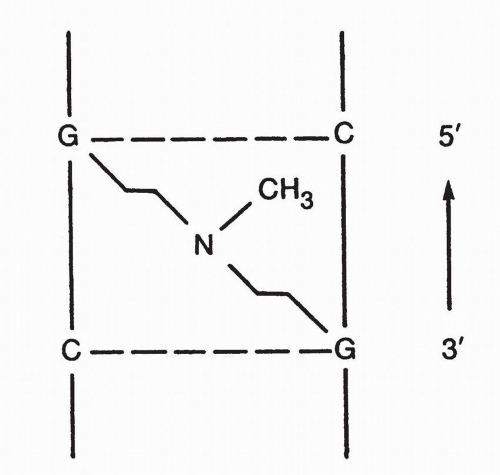
Data suggest that alkylation is nonuniform along the DNA strand and may be concentrated in specific regions. One determinant of regional specificity of DNA alkylations may be chromatin structure
11,
83; areas of active transcription seem to be most vulnerable. Additionally, evidence shows that nitrogen mustards such as mechlorethamine preferentially cross-link at 5′-GNC sequence.
84In summary, the preponderance of evidence supports the hypothesis that the major factor in the cytotoxicity of most clinically effective alkylating agents is interstrand DNA cross-linking, which results in inactivation of the DNA template, cessation of DNA synthesis, and ultimately cell death. Cell-cycle checkpoint proteins, including most prominently p53, are responsible for the recognition of DNA alkylation and strand breaks. Recognition of DNA damage leads to a halt in cell-cycle progression and initiation of programmed cell death. Cells containing mutated p53 have greater resistance to alkylating agents.
85An increased knowledge of alkylation mechanisms and targets may make it possible to improve the therapeutic index of these agents. For example, the therapeutic index of alkylating agents should improve if the alkylation of tumor cells were increased without a simultaneous increase in normal tissue alkylation. This might be accomplished by maneuvers designed to inhibit drug activation by glutathione or GST P1,
86 or by blocking the repair processes, as discussed below.
Tumor Resistance
The emergence of alkylating agent-resistant tumor cells is a major problem that limits the clinical effectiveness of these drugs. Cellular resistance mechanisms identified in preclinical experimental settings include drug uptake, enhanced anti-apoptosis pathways, activation of survival pathways, enhanced intratumoral drug inactivation, and changes in DNA repair.
Decreased Cellular Uptake of Selected Alkylating Agents
Several of the drugs (melphalan, nitrogen mustard) of this class require active transport into cells. One mechanism for drug resistance is decreased drug entry into the cell. This mechanism was best demonstrated in L5178Y lymphoblast cells resistant to mechlorethamine.
56 The extracellular domain of the leucine-melphalan transporter expresses CD98. Reduced expression of CD98 on human myeloma cells is associated with melphalan resistance.
87 The glutathione-dependent efflux transporters MRP1 and MRP2 can confer resistance to chlorambucil.
89
Resistance due to Inactivation by Glutathione or GSTs
Intracellular inactivation of alkylating agents has been implicated in human tumor resistance. Early studies showed increased levels of sulfhydryls associated with resistance in experimental tumors, and increased nonprotein sulfhydryl content, particularly in the form of glutathione, in resistant tumor cell lines.
88 While increased intracellular glutathione content may be found in resistant cells, elevated GST activity may also play a role
90 and increased aldehyde dehydrogenase (ALDH) activity, which converts aldophosphamide to the inactive carboxyphosphamide, was present in cells resistant to cyclophosphamide.
91,
92,
93Alterations in the GSH/GST system found in alkylating agent resistance phenotypes include increased intracellular GSH levels, elevation of GST activity, and changes in the expressed levels of one or more GST isozymes. Currently, several GSH-related mechanisms may explain the observed tumor cell resistance to alkylating agents, including (a) enhanced inactivation of electrophilic alkylating agents, such as melphalan
94 by direct conjugation to GSH; (b) GSHdependent denitrosation of nitrosoureas, a reaction that is preferentially catalyzed by one of the rat liver GST μ enzymes in the case of BCNU; (c) scavenging for reactive organic peroxidases, a process that is catalyzed by GSH peroxidase;
95 and (d) quenching of chloroethylated-DNA monoaducts.
96Inherited polymorphisms of functional significance have been reported in genes that encode glutathione-
S-transferases (GSTs) and may contribute to resistance (see
Chapter 6). There are four cytosolic families of GSTs, including GST
α, GST
μ, GST
θ, and GST
π.97 Gene clusters of GST
μ (
GSTM1, M2, M3, M4, and
M5) and GST
θ (
GSTT1 and
T2) are located on chromosomes 1 and 22, respectively.
98 Independent gene deletions at
GSTM1 and
GSTT1 loci result in a lack of active protein in ≈50% and 20% of Caucasians, respectively.
99 GST
π or GSTP1, encoded by a single locus (
GSTP1) on chromosome 11, is also subject to polymorphic variation.
100 Codon 105 residue forms part of the GSTP1 active site for binding of hydrophobic electrophiles,
101 and the Ile-Val substitution affects substrate-specific catalytic activity and thermal stability of the encoded protein.
102 Reactive metabolites of ifosfamide, busulfan, and chlorambucil are substrates for GSTP1-mediated
glutathione conjugation in vitro. Allelic variants of GSTP1 differ significantly in their efficacy in catalyzing the GSH conjugation and hence their ability to detoxify alkylating agents.
103 The effect of these polymorphisms on clinical conjugation, toxicity, and antitumor response is uncertain, and the studies to date are summarized in
Chapter 6.
Resistance to Cyclophosphamide due to Elevated Aldehyde Dehydrogenase Activity
Resistance to cyclophosphamide may also be determined by the activity of cellular ALDH.
93,
104 ALDH is an enzyme responsible for the oxidation of intracellular aldehydes.
105 The cytoplasmatic ALDH isozyme converts activated cyclophosphamide to the inactive excretory product, carboxyphosphamide, in both murine and human cell lines.
106 There is no clear evidence that ALDH activity confers resistance in human tumors.
ALDH may have an important role in early differentiation of hematopoietic stem cells by oxidizing retinol to retinoic acid.
107 It is hypothesized that cancer stem cells survive cyclophosphamide treatment due to ALDH. Murine and human hematopoietic and neural stem and progenitor cells have high ALDH activity.
108,
109 Increased ALDH activity has also been found in malignant stem cell populations in multiple myeloma and acute myeloid leukemia.
110
DNA repair and Alkylating Agent Resistance
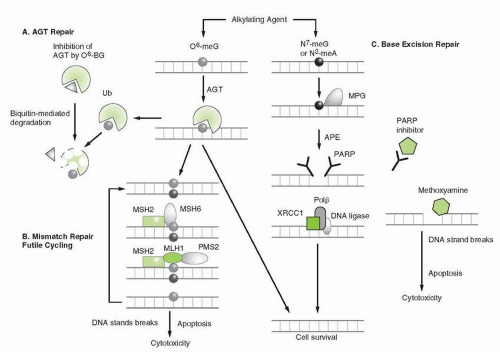
Enhanced repair of DNA lesions generated by alkylation plays a clearly established role in resistance of experimental and human tumor cells to alkylating agents. Because DNA appears to be the most critical target for the alkylating agents, its repair has been a major focus of study and several mechanisms involved in repairing alkylation and strand breaks are summarized in
Figure 14A-8.
Enhanced excision of alkylated nucleotides from DNA as a mechanism of resistance to alkylating agents was first demonstrated in bacteria
75 and later in mammalian cells. Bacterial, fungal, and mammalian cells are capable of excising and repairing sites of alkylation, as well as removing cross-links and repairing single- and double-strand breaks.
Alkylguanine DNA Alkyltransferase Mediated repair
Repair of DNA alkylation products and cross-links involves multiple systems, each composed of one or several distinct enzymes
(see
Fig. 14A-8). The simplest of these catalyzes the transfer of alkyl substituents (methyl-, ethyl-, benzyl-, 2-chloroethyl-, and pyridyloxobutyl-) from the
O6-position of guanine to an active cysteine acceptor site within the protein in a single enzyme repair process. This enzyme, alkylguanine-
O6-alkyl transferase (AGT), is encoded by the MGMT (
O6-methylguanine methyltransferase) gene in humans and is homologous to the bacterial alkyltransferase gene, ada.
Several clinical trials have established an inverse correlation between AGT content in brain tumors and the response to treatment of brain tumor patients receiving BCNU, temozolomide, and other alkylating agents.
111,
112,
113 Lower levels of AGT activity result from epigenetic silencing due to MGMT promoter methylation. Tumors with methylated MGMT are highly responsive to these agents, while those with fully expressed AGT tend to be resistant. The availability of 5′ cytosine methylation-specific PCR (MSP) provides a facile assessment of MGMT promoter methylation and has value as a predictive assay for response to alkylating agent-based chemotherapy. MGMT promoter methylation has been correlated with survival in patients with glioma treated with nitrosoureas and temozolomide
82,
114 and prolonged progression-free survival (PFS) in patients treated with temozolomide.
115,
116 Together, these data illustrate a role for MGMT gene function in glioma chemoresistance to alkylating agents. Clinical trials attempting to modulate AGT activity are discussed below.
111,
117,
118
Mismatch Repair
A second DNA repair system, mismatch repair (MMR), recognizes the mismatch created by alkylation of DNA bases, and, after unsuccessful attempts at repair, triggers cell-cycle arrest and apoptosis. For example,
O6 alkylation of guanine leads to mispairing of the damaged base with thymine, creating a distortion in the DNA double helix, which is recognized by components of the MMR complex. MMMR deficiency has been associated with resistance to alkylating agents owing to the inability to recognize the mismatch and initiate the cycle of futile repair attempts, cell-cycle arrest, and apoptosis.
119,
120 Several proteins comprise the MMR pathway (hMLH1, hPMS2, hMSH2, hMSH3, and hMSH6), which is programmed to correct erroneous DNA base pairing.
In MMR competent cells during DNA replication, DNA polymerase mispairs unresolved
O6-methylguanine with thymine. The mismatch triggers attempts to remove the mispaired thymine. If repair is successful, subsequent rounds of replication continue to mispair
O6-methylguanine with thymine, resulting in repetitive futile cycles of MMR. This futile cycle may induce double-strand breaks, which in turn, trigger p53-dependent cell-cycle arrest and apoptosis.
121 Loss of MMR competence creates tolerance to the mispaired bases and allows cell replication and survival, provided the DNA lesions are compatible with viability. The loss of MLH6 is a frequent event in glioma cells selected by resistance to temozolomide.
122Specific interactions of MMR proteins with cell-cycle checkpoint proteins have been implicated in apoptosis. hMLH1 has been associated with signaling ATR-dependent G2 cell-cycle arrest in response to DNA methylation,
123 and MMR proteins may initiate degradation of cyclin D1 following alkylation to promote cell-cycle arrest in response to alkylation.
124 Functional MSH2 and hMLH1 are also believed to activate p73-dependent apoptosis pathways via c-Abl.
125Resistance to alkylating agents conferred by MMR deficiency is further illustrated by hereditary nonpolyposis colon cancer, which is caused by mutations in hMLH1 or hMSH2 genes. Colon cancer cells harboring these mutations are resistant to alkylating agents,
126 as are other cell lines deficient in hMLH1.
127
DNA Excision Repair
NA excision repair pathways provide a comprehensive mechanism for recognizing and removing damaged bases or nucleotide segments from a single DNA strand and then resynthesizing the new DNA segment, using the opposing undamaged strand as a template. Excision repair complexes includes base excision repair (BER) and nucleotide excision repair (NER).
Base Excision Repair
In response to DNA alkylation, BER is initiated by the action of damage-specific DNA glycosylases that recognize and excise single base lesions such as N
3 methyladenine and N
7-methylguanine. Release of the damaged base produces an apurinic/apyrimidinic (AP) site, which is excised by the APE endonuclease (
Fig. 14A-9). The missing segment is then resynthesized by DNA polymerase and sealed by DNA ligase. Persistent AP sites are recognized by topoisomerase I and II and these may form cleavable complexes that induce apoptotic signals. The enzyme PARP plays a pivotal role in the recognition of strand breaks and in the formation of DNA strand break intermediates that attract repair complexes. The N
3-methyladenine and N
7-methylguanine are the most common adducts created by alkylation and account for greater than 80% of all methylation events. However, these lesions contribute modestly to the cytotoxicity of alkylating agents due to the efficiency of the BER pathway. Perturbation of BER capacity through alterations of glycosylase expression or through pathway inhibition greatly decreases the efficiency of N3 and N
7 methyl adduct repair.
128 Targeting of BER, as described below, is particularly effective in enhancing the sensitivity to platinating agents in various cell lines tumors, especially breast cancers deficient in BRCA 1 and BRCA 2, and to alkylating agents in MMR-deficient tumors.
129
Nucleotide Excision Repair
NER is an additional mechanism for excising bulky alkylation products and DNA intrastrand cross-links. The pathway includes multiple proteins that recognize DNA adducts, such as those produced by alkyl lesions and incise 3′ and 5′ to the damaged base(s), causing release of the damaged nucleotides and surrounding segments of DNA. Excision is followed by resynthesis of the missing segment, using the opposing strand as a template. Components of the NER complex also have a role in repair of double-strand breaks. NER-deficient mammalian cells, such as those derived form patients with xeroderma pigmentosum (XP), are hypersensitive to alkylating and cross-linking agents.
130,
131 Studies of the effects of NER deficiency
on the toxicity of alkylating agents are most extensive in rodent models, but there is evidence that polymorphic variants of ERCC1 confer increased alkylating agent sensitivity for both normal and malignant tissues, influence response to treatment, and greater toxicity.
132 The role of NER in alkylating agent sensitivity and resistance in clinical cancer treatment is under active investigation.
Cross-Link Repair
Interstrand cross-links covalently tether strands of DNA, preventing unwinding of duplex DNA and prohibiting polymerase access. Both strands of DNA are involved in this lesion, precluding straightforward excision repair and gap-filling pathways. Consequently, the repair of interstrand DNA cross-links is complex, integrating elements of the NER pathway, a variety of less well-understood activities to form a double-strand break, insertion of new bases, and homologous recombination (HR). Though mechanisms are incompletely understood several, mammalian cell types have extreme sensitivity to cross-linking agents. Faconi anemia
133 and Bloom’s (BLM) syndrome cells
134 are both hypersensitive to alkylating agents that cause interstrand cross-links. The Fanconi anemia pathway and the BLM helicase are believed to be activated in response to replication stalling due to cross-linked DNA; their dysfunction in the inherited disorders accounts for alkylating agent sensitivity.
135,
136 Furthermore, mutations in excision repair genes ERCC1 and ERCC4 (XPF) also render cells sensitive to cross-linking by alkylating agents, suggesting these genes play a role in the repair of cross-links in addition to their role in NER.
137
Akt
The Akt family in humans is comprised of three genes (Akt1, Akt2, and Akt3) encoding for serine/threonine protein kinases (PKB). Akt activation occurs downstream of various receptor tyrosine kinases and phosphatidylinositol 3-kinase (PI3K). The PI3K/Akt pathway is frequently activated in human cancer and has been implicated in tumor cell proliferation, cellular survival, and chemotherapy resistance (see
Chapter 30). In response to alkylation by the agent temozolomide, Akt is induced in lymphoblastoid, colon, and breast cancer cells in a mismatch repair-dependent manner.
138,
139Upon activation, PI3K/Akt signaling confers resistance to chemotherapy through its antiapoptotic effects as mediated by phosphorylation of several downstream targets. Activated Akt is thought to inhibit apoptosis by phosphorylating molecules upstream and downstream of the mitochondrial apoptotic pathway. Akt-dependent phosphorylation of proapoptotic BH3 family members such as Bad, Bax and Bim-EL decreases the ability of these proteins to hold mitochondria in an open configuration, resulting in a reduction in cytochrome c release. Akt can phosphorylate caspase-9 to inhibit its ability to activate executioner caspase 3.
140,
141 Akt reduces cytochrome c release through modification of the antiapoptotic Bcl-2 homologous Mcl-1.
142,
143 Akt exerts indirect inhibition of apoptosis through effects on p53, the most important regulator of apoptosis.
144Akt also drives chemoresistance by promoting cell growth. Akt is involved in the survival pathway of mammalian target of rapamycin (mTOR), a serine/threonine kinase that is implicated in protein synthesis control.
145 Akt activates mTOR complex 1 (mTORC1; or mTOR-raptor complex) indirectly by inhibiting phosphorylation of tuberous sclerosis complex 2, thereby allowing Ras-related small G protein (Rheb)-GTP to activate mTORC1 signaling.
146
Defects in Cell Cycle Arrest and Apoptosis
In addition to the mechanisms described above, mutation or silencing of genes that induces cell-cycle arrest and apoptosis may lead to alkylator resistance.
In cells that experience genotoxic stress during replication, activation of these factors triggers the signaling cascades, leading to delayed progression through S-phase in order to allow time for DNA repair.
147 In the presence of DNA damage, an S-phase cell-cycle checkpoint is activated by the checkpoint kinases ataxia-telangectasia mutated (ATM) and ATM- and Rad3-related (ATR). Activation depends on the type of DNA damage: ATM is recruited to DNA double-strand breaks (DSBs) induced by agents such as ionizing radiation (IR), whereas ATR is recruited to sites of replication protein A (RPA)-coated single-stranded DNA (ssDNA). These sites accumulate at stalled replication forks or at sites of single-strand damage.
148,
149 The involvement of ATM and ATR in the response to carcinogen-induced DNA damage has been established,
150 by showing an enhanced sensitivity of ATM- and ATR-defective cells to methylating agents.
Two parallel branches of the DNA damage-dependent S-phase checkpoint are thought to cooperate by inhibiting distinct steps of DNA replication. One branch is activated by the phosphorylation of structural maintenance of chromosomes 1 (SMC1), a cohesin that is activated by ATM or ATR.
151 The second branch, consisting of the ATR-Chk1- or ATM-Chk2-complexes, regulates turnover of CDC25A, a phosphatase that regulates Cdk2 and consequently blocks the initiation of replication.
152Defects in damage recognition or apoptotic signaling may lead to relative resistance.
153 For example, loss of normal p53 function, up-regulation of the antiapoptotic proteins, Bcl-2 or Bcl-X
L, or overexpression of the epidermal-growth factor receptor (EGFR) can disrupt the normal apoptotic response to DNA damage caused by alkylating agents.
154,
155 As mentioned previously, apoptotic cell death after DNA damage is mediated through p53, which blocks cell-cycle progression, initiates attempts to repair damage, and ultimately activates apoptotic pathways.