The acquired immunodeficiency syndrome (AIDS) epidemic is in its third decade, and the number of people infected with the human immunodeficiency virus-1 (HIV) in the United States is approaching 1 million, with nearly 40 million individuals infected worldwide. Since early in the HIV epidemic it has been recognized that certain tumors occur with increased frequency in the setting of HIV-induced immunosuppression. These include most prominently Kaposi sarcoma (KS), B-cell non-Hodgkin lymphoma (NHL), and anogenital neoplasia. Less common, but also of increased frequency compared with the general population, are Hodgkin lymphomas and, in children, leiomyosarcomas. The advent of highly active antiretroviral therapy (HAART) has markedly reduced the morbidity and mortality of HIV-infected individuals with access to such medications. Oncologic complications are among those markedly reduced in the era of better viral control, although the impact has not been uniform across neoplasms.
Pathogenesis of AIDS-Related Neoplasms
Several possible contributing pathophysiologic mechanisms probably participate in the predisposition to neoplasms in the setting of HIV infection, including altered immune activation, impaired innate tumor surveillance, dysregulated cytokine production, and inadequate control of oncogenic pathogens. Such issues are not unique to HIV-related immunosuppression and are analogous in certain respects to malignancy in patients with congenital or therapeutic immune deficiency states, although there are clear differences among these entities.
Organ transplant data indicate that the risk of post-transplant lymphoproliferative disease increases proportionately with the depth and duration of therapeutic immunosuppression (see Table 19.1 ). There are also data suggesting that immune activation may contribute to the emergence of lymphoid neoplasms. There is an increased incidence of lymphomas in the setting of autoimmune diseases such as Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, and celiac disease. Similarly, immune activation may be directly driven by an infectious agent. Inadequate host immunologic responses to infectious agents may result in tumor development due to a combination of impaired immunity and viral oncogenesis. The geographic clustering of KS and its association with specific sexual practices strongly suggested an association with a secondary infectious process, a process subsequently identified as human herpesvirus-8 (HHV-8) or KS herpesvirus, a member of the γ-herpesvirus family. The common association between KS and the polyclonal lymphoproliferative disorder multicentric Castleman disease led to the identification of HHV-8 as the driving pathogen of that disorder, and it has subsequently been implicated in the pathogenesis of primary effusion lymphoma, as well. The role for immune control of HHV-8 and Epstein-Barr virus (EBV) in oncogenesis is now strongly supported by evidence that improved immune function with HAART markedly reduces HHV-8- and EBV-related tumors. Notably, however, data regarding the impact of HAART on human papillomavirus (HPV)-related tumors are less clear, and it is not yet known to what extent improved immune function will affect anogenital neoplasia. What is clear is that HIV itself does not play a direct role in tumor generation. Other than in very rare cases of T-cell lymphoma, HIV is not detectable in HIV-related malignancies. HIV provides the immunologic dysfunction permissive of tumor emergence. These tumors may be regarded as opportunistic neoplasms in much the same way as specific infections are regarded as opportunistic in the immunocompromised host.
| Transplant Type | Incidence (%) * |
|---|---|
| Renal | 1.0 |
| Heart | 1.8 |
| Liver | 2.2 |
| Heart and lung | 5.0 |
| Bone marrow (BM) | <1.0 |
| T cell–depleted BM | 12.0 |
| Mismatched, T cell–depleted BM | 24.0 |
* Incidence figures would be expected to be lower using current protocols.
Perturbations in the tissue cytokine milieu may provide the specific signals altering the control of cellular proliferation in tumors of immune suppression. KS serves as an example of this, in that KS cells, unlike normal mesenchymal cells, both secrete interleukin-6 (IL-6) and have a proliferative response to IL-6 in an autocrine fashion. Similarly, some AIDS-lymphoma cells appear to produce IL-10, and their growth rate is affected by it. These events may not be sufficient for tumor generation but provide the proliferative background against which transforming events may occur.
Kaposi Sarcoma
KS has been the most common HIV-related neoplasm, although the incidence has declined significantly since the introduction of HAART. It is believed to arise from mesodermally derived cells, the exact nature of which remains controversial. Histologically these lesions are composed of multiple cell types, including smooth muscle, endothelial, and immune cells. The histologic picture raises the unanswered question of whether this disease represents a true malignancy or is the result of dysregulated proliferation of otherwise normal cells in response to an abnormal signal. Efforts to define clonality in lesions have conflicting results, showing that clonal disease may evolve, but polyclonality is common. The abnormal drive for cell proliferation may be due to products of KS herpesvirus itself such as a constitutively active G protein–coupled receptor the virus encodes. In addition, the HIV gene product, tat , may induce the lytic phase of HHV-8, resulting in increased viral transcripts, IL-6 production, and stimulation of the JAK/STAT proliferation pathway, which may account for the extraordinary predilection for KS seen in HIV-infected individuals above that of other immunosuppressed populations. The epidemiology of KS is outlined in Figures 19.1 and 19.2 .


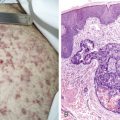
KS lesions occur clinically in the skin, on mucosal surfaces, in lymph nodes, and in solid organs, most commonly the lung and gastrointestinal (GI) tract. Cutaneous manifestations vary from erythematous, macular lesions to raised, nodular masses with a violaceous hue. Large, plaquelike lesions may develop when clusters of tumors coalesce, and central necrosis can occur. KS tends to appear at multiple sites concurrently, with no ordered pattern of spread. An important differential diagnosis is bacillary angiomatosis, which is an infectious disease occurring in HIV patients caused by Bartonella organisms. Cutaneous Pneumocystis carinii infection has also been reported to cause an erythematous lesion resembling KS. Early KS lesions may also be mistaken for benign skin lesions such as dermatofibromas or angiomas. Thus, biopsy of clinically suspected KS lesions is recommended at the time of first clinical presentation.
Mucosal involvement by KS may occur in the oral cavity, conjunctiva, or more rarely the urethral meatus and may result in local discomfort. Cutaneous lesions, however, are mostly of cosmetic significance, and their successful treatment can overcome a major source of distress in affected patients. Approximately one half of patients with mucocutaneous disease will also have KS involving other organs. While skin disease usually accompanies organ involvement, up to 15% of patients have been reported to have lymph node, GI, or lung KS without skin manifestations.
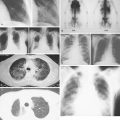
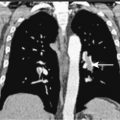
Pulmonary KS may involve the large airways, interstitium, alveoli, or pleural surfaces, and the clinical features vary accordingly. Patients may complain of dyspnea, cough, hemoptysis, or wheezing, but the disease is often asymptomatic. When parenchymal involvement is extensive it may be life-threatening, prompting most clinicians to aggressively treat patients in whom infiltrates on chest radiography are thought to be due to KS. GI tract lesions are generally asymptomatic but may result in nonspecific symptoms such as abdominal pain and bloating. These lesions may be the cause of minor chronic blood loss, but massive hemorrhage is uncommon.
Finally, KS can involve lymph nodes and local lymphatics, causing marked local edema. This is exacerbated by the in- creased permeability of the vascular component of KS and by a permeability factor elaborated by KS cells, vascular endothelial growth factor. Edema is commonly seen in the groin, distal lower extremities, or head and neck regions, and is a major cause of morbidity. It is the most common cause of symptoms from KS and is generally regarded by us as an indication to proceed with a more aggressive treatment approach. A system for clinically staging KS is outlined in Table 19.2 .
| Good Risk (All of the Following) | Poor Risk (Any of the Following) | |
|---|---|---|
| Tumor (T) | Confined to skin and/or lymph nodes and/or minimal oral disease | Tumor-associated edema or ulceration |
| Extensive KS | ||
| Gastrointestinal disease | ||
| Other visceral involvement | ||
| Immune system (I) | CD4 > 200 cells/μL | CD4 <200 cells/μL |
| Systemic illness (S) | No opportunistic infections or thrush (Candida) | History of or current opportunistic infection or thrush“ |
| No “B” symptoms | B” symptoms | |
| Karnofsky score >70% | Karnofsky score <70%Other HIV-related illness(CMV, PCP, MAI, PML) |
KS is not curable with standard therapies, and so goals of treatment are to prevent deleterious consequences of the disease, including organ dysfunction from extracutaneous involvement as well as discomfort or cosmetics associated with skin lesions. Initiation of HAART often results in regression of KS lesions, and so antiretroviral therapy should be considered in all patients. It should be noted, however, that rarely, rapid progression of KS has been reported after HAART initiation as a component of the immune reconstitution inflammatory syndrome. For local control, topical approaches include topical retinoids, local radiation therapy, cryotherapy, laser therapy, and direct intralesional injections of chemotherapy. Treatment of advanced disease most commonly consists of single-agent chemotherapy, with liposomal doxorubicin, paclitaxel, vinca alkaloids, or etoposide. Novel treatments that have shown early promise and are the subject of ongoing clinical trials include angiogenesis inhibitors, mTOR inhibitors, and the tyrosine kinase inhibitor imatinib mesylate, among others.
Non-Hodgkin Lymphoma
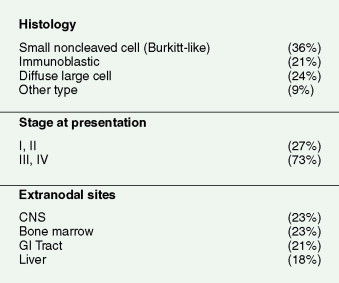
The increased incidence of NHL in AIDS patients was first noted in 1984; NHL became recognized as an AIDS-defining illness in 1987. The estimated incidence of NHL in patients ranges from 1.6% to 2.0% per year, with patients from all HIV risk groups being susceptible to the disease. AIDS-related NHLs are a heterogeneous group in terms of pathogenesis, histologic appearance, clinical behavior, and treatment. Diffuse large B-cell lymphoma (DLBCL) is the most common, particularly the immunoblastic variant. DLBCL may be limited to the brain and leptomeninges, in which case it is referred to as the distinct entity of primary central nervous system (CNS) lymphoma. Burkitt lymphoma is the second most common systemic HIV-related lymphoma and is the only AIDS-related NHL that customarily occurs with CD4 counts over 200, and thus, not surprisingly, is the only AIDS-related NHL not to see a decrease in incidence since the introduction of HAART. Rare AIDS-related lymphomas occurring only at very low CD4 counts are plasmablastic lymphoma and primary effusion lymphoma, also known as body cavity lymphoma.
The pathogenesis of AIDS-related lymphoma is related to underlying immune dysfunction, stimulation of B cells by abnormally produced cytokines, and co-infecting oncogenic organisms such as EBV and HHV-8. DLBCLs are associated with EBV in up to 75% of cases and in virtually all primary CNS lymphomas, while Burkitt lymphomas are EBV-associated in only 20% to 35%. Interestingly, the endemic form of Burkitt lymphoma, traditionally presenting as jaw tumors in African children, is uniformly EBV-positive, while traditional nonendemic Burkitt lymphomas in non-HIV-infected patients are uniformly EBV-negative. The pattern of EBV expression in AIDS-related lymphomas differs from that seen in post-transplant lymphoproliferative disease. In systemic AIDS lymphomas a unique combination of Epstein-Barr nuclear antigen-1 and latent membrane protein-1 expression occurs. In post-transplant patients and primary CNS AIDS lymphomas, Epstein-Barr nuclear antigen-2 through -5 are expressed along with latent membrane protein-1 and -2. Transplant-related neoplasms may regress upon withdrawal of administered immunosuppressive agents or occasionally with antiviral therapy. However, this is not the case in AIDS, where tumor regression is rare even in the face of improved immune function with HAART.
Several genetic abnormalities have been found in AIDS-related NHL. Rearrangements of the c- MYC proto-oncogene located on chromosome 8 with the immunoglobulin genes situated on chromosomes 14 and 2 are the most common mutations noted, with a frequency of 23% to 79%. The rearrangement occurs in the mature B cell, in which immunoglobulin gene rearrangement has already occurred. The resultant juxtaposition of the c- MYC and immunoglobulin genes may play a central role in the transformation of cells bearing this molecular translocation. Other oncogene mutations that occur less frequently than that involving c- MYC include abnormalities of the TP53 tumor suppressor gene and the RAS oncogene. EBV is implicated in the pathogenesis of virtually all cases of plasmablastic lymphoma, a highly aggressive B-cell lymphoma often presenting in the oral cavity. HHV-8 underlies the rare cases of primary effusion lymphoma, which occurs without accompanying nodal disease in serosal sites such as the peritoneum, pleura, and pericardium. EBV co-infection is also common. Plasmablastic and primary effusion lymphomas predominantly occur with CD4 counts less than 50.
Histologically, AIDS-related NHLs are high-grade B-cell malignancies demonstrating aggressive clinical behavior. Rarely, T-cell malignancies such as large granular lymphoproliferative disease, Sézary syndrome, or angioimmunoblastic T-cell lymphoma are encountered in HIV-infected patients. In some cases these rare T-cell malignancies have been demonstrated to harbor HIV, some of which have integrated into the host genome resulting in a transforming mutation.
The clinical features of AIDS-related NHL are similar to those of aggressive lymphomas in general. Patients with AIDS-related lymphomas are, however, more likely to present with advanced-stage disease and involvement of multiple extranodal sites, as compared with their HIV-negative counterparts. There is also a high proportion of patients with systemic “B” symptoms of significant weight loss, fevers, and drenching night sweats (see Fig. 19.3 ). Patients presenting with such symptoms should have a thorough microbiologic evaluation to exclude bacterial, mycobacterial, viral, fungal, or parasitic disease. HHV-8–associated multicentric Castleman disease should also be considered in the differential diagnosis of AIDS-related lymphoma, because it presents similarly with diffuse adenopathy and profound “B” symptoms and often precedes the development of, or presents concurrently with, AIDS-related lymphoma.
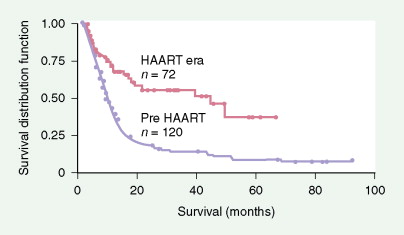
Prognosis is closely linked to the status of the immunodeficiency (as indicated by CD4 count and prior AIDS diagnosis) as well as tumor-related factors. Prognosis has improved significantly since the introduction of HAART, with the median survival in AIDS-related DLBCL increasing from 8 months to 43 months (see Fig. 19.4 ). High-risk international prognostic index score is a poor prognostic factor in AIDS-related lymphoma, as it is in non-AIDS-related lymphoma. Other poor prognostic factors include Burkitt, plasmablastic, or primary effusion histologies, CD4 count less than 100 cells/μL, and failure to achieve a complete remission to initial therapy. When one is treating patients with AIDS-related lymphoma, the status of the HIV disease itself must always be kept in mind, since prognosis and tolerance of therapy are different for patients with advanced AIDS failing antiretrovirals compared with those tolerating and benefiting from HAART.
Primary CNS lymphoma is an important subgroup of HIV-associated lymphoma and composes 15% to 20% of all AIDS NHLs. It tends to occur in patients with more advanced immunosuppression; one study demonstrated a mean CD4 count of 30 cells/mm 3 in primary CNS lymphoma versus 190 cells/mm 3 in patients with systemic NHL. Primary CNS lymphoma in AIDS resembles transplant lymphoma in that it is of immunoblastic histology and is virtually always associated with the EBV genome (see Table 19.3 ). This is in contrast to systemic HIV lymphoma, which differs from lymphoma developing in patients who are immunocompromised for other reasons (see Table 19.4 ). This subset of HIV-associated lymphomas has substantially declined in the era of HAART. The improved immune function associated with more effective control of HIV probably accounts for the decrease in patients with this devastating complication. Clinical presentation is with neurologic symptoms which may be as vague as subtle personality changes with a CNS mass lesion found by radiographic imaging studies. In AIDS patients, lymphoma must be distinguished from Toxoplasma or other infectious brain abscesses, as well as progressive multifocal leukoencephalopathy. Certain radiologic criteria are useful in differentiating lymphoma from Toxoplasma lesions: NHL is typically located centrally, is often larger than 2 cm, and may cross the midline, while converse features may suggest toxoplasmosis. Brain biopsy is required to establish a definitive diagnosis, but the presence of EBV by cerebrospinal fluid (CSF) polymerase chain reaction strongly supports a lymphoma diagnosis and has a sensitivity and specificity of greater than 90% in patients with a negative Toxoplasma titer who have been on trimethoprim-sulfamethoxazole prophylaxis. Analysis of the CSF for a clonal IgH gene rearrangement by polymerase chain reaction is also highly suggestive of CNS lymphoma, while routine cytologic evaluation and flow cytometry are much less sensitive as a result of the paucity of circulating cells. For those in whom ambiguity remains, a therapeutic trial of antitoxoplasmosis therapy may be useful, since a majority of patients with Toxoplasma infection will respond within 2 weeks.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


