INTRODUCTION
It is clinically useful to divide non-Hodgkin lymphomas (NHL) into indolent and aggressive based on their clinical behavior (1). Patients with indolent NHL typically have a survival of many years, even if untreated, but paradoxically are usually incurable. Patients with aggressive NHL have a survival time measured in weeks to months if untreated yet are usually chemosensitive and frequently curable. In this chapter, we focus on the clinical characteristics, pathology, and treatment of aggressive NHL.
EPIDEMIOLOGY
The incidence of NHL has increased over the last five decades, as reported by US and international registries (2,3,4). During the years 1993 to 1995, the age-adjusted incidence increased 3% per year according to data from the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute (2). Some of this increased incidence can be attributed to the acquired immunodeficiency syndrome (AIDS), but this epidemic does not explain the increase of NHL before 1980. In the elderly population, there has also been a marked increase of NHL, largely the indolent NHLs, which are discussed in Chapter 7.
An estimated 71,850 new cases of NHL will be diagnosed in the United States in 2015, and 19,790 NHL-related deaths will occur. In 2014, NHL was the ninth largest cause of death among men and the eighth largest cause in women (3% of all cancer-related deaths). Diffuse large B-cell lymphoma (DLBCL), the most common NHL subtype, has an aggressive behavior and is more common in whites than African Americans in the United States; however, 5-year survival outcomes are worse in African Americans (5).
ETIOLOGY
Most cases of aggressive NHL do not have a well-defined cause. Recent work has shown that the lifetime risk for many cancers correlates with the total number of divisions of normal self-renewing cells (6). The implications of these findings suggest that many cancers may not be due to hereditable genetic aberrancies, environmental exposures, infectious etiologies, or other known causes, but instead are attributable to a combination of factors that may include chance. For the NHLs that appear to have currently identifiable drivers, there are four groups of drivers: immune suppression (both acquired and primary), infectious agents, toxic exposure, and familial (Table 8-1).
Inherited and acquired immune deficiency Wiskott-Aldrich syndrome Ataxia-telangiectasia Chédiak-Higashi syndrome X-linked immunoproliferative disorder Severe combined immunodeficiency Common variable immune deficiency Iatrogenic immune suppression Solid organ or bone marrow transplant |
Toxic exposures Prior chemotherapy Phenoxyherbicides Dioxin Radiation or radiation therapy |
Infectious exposures Epstein-Barr virus Human T-cell leukemia/lymphoma virus Human herpesvirus type 8 (HHV-8) Human immunodeficiency virus (HIV) |
Autoimmune disorders Sjögren syndrome Celiac sprue Systemic lupus erythematosus Rheumatoid arthritis |
The strongest association is with immune suppression, both primary and acquired (7). Examples of primary immunodeficiency include inherited immune disorders, such as Wiskott-Aldrich syndrome, severe combined immune deficiency, common variable immune deficiency, and ataxia-telangiectasia (8). These and other inherited disorders are associated with an increased lifetime risk of developing NHL, with aggressive B-cell NHL being most common.
Patients who are immunosuppressed for therapeutic reasons—for example, after transplantation—are also at increased risk of NHL, especially if treated with cyclosporine, azathioprine, prednisone, or monoclonal antibodies for the removal of T cells (9). A loose association can be drawn between the level of immune suppression and lymphoma risk. Transplant patients treated with the highest doses of immunosuppressive agents, such as heart transplant recipients, are at greater risk of developing lymphomas. These lesions are also more likely to be aggressive, extranodal forms. Individuals treated with pharmacologic immune suppression for autoimmune disorders—such as systemic lupus erythematosus, Sjögren syndrome, or rheumatoid arthritis—are also at increased risk for NHL, including the tumor necrosis factor antagonists (10). A subset of these NHLs is histologically aggressive and associated with Epstein-Barr virus (EBV). These lesions may regress following withdrawal of the immunosuppressive agent, speaking to the complex interaction between the immune system and an immune clonal population (11).
Infectious agents associated with development of aggressive NHL include human immunodeficiency virus (HIV), EBV, human herpesvirus type 8, and human T-cell lymphotropic virus type 1 (12). The greatest factor involved in the worldwide increase in NHL, although lessened with the advent of highly active antiretroviral therapy (HAART), is HIV infection (13). The risk of NHL is increased by up to 300% in untreated HIV-infected patients, rising in proportion to the duration of the HIV infection. Although the risk of NHL in HIV-infected patients appears to be decreased by HAART, the relative risk of NHL remains much higher than that for those not infected with HIV. Aggressive NHL can occur in HIV-infected patients at any stage of infection, but the risk increases as CD4 counts drop to <100 × 103/μL, and NHL is considered an AIDS-defining illness.
EBV also plays a role in lymphomagenesis, due in part to chronic antigenic stimulation (14). EBV is virtually always associated with certain types of NHL, such as endemic (African) Burkitt lymphoma (BL) and extranodal T-cell/natural killer (NK)-cell lymphoma of nasal type, with many other NHL subtypes occasionally involved. Many patients with HIV-related lymphomas are co-infected by EBV, including HIV-associated primary central nervous system (CNS) lymphoma, which is infected in essentially 100% of cases (15). Human herpesvirus type 8 is associated with primary effusion lymphoma (PEL), which tends to occur in HIV-infected patients. Human T-cell lymphotropic virus type 1 is associated with adult T-cell lymphoma/leukemia.
Environmental and occupational exposures to toxins are associated with an increased risk of NHL (16). Herbicides, especially phenoxyacetic acid derivatives, are associated with NHL, especially in the farming belts of the United States. Occupations held by individuals reported to be at increased risk for NHL include farming, metalworking, forestry, aircraft maintenance, woodworking, and dry cleaning. One of the common exposures in these industries is the use of organic solvents.
Family history of NHL is also a potential risk factor for some lymphomas. Individuals who have relatives with NHL may have a slightly higher risk of developing NHL, but data are inconclusive and mechanisms are unclear (17).
CLINICAL PRESENTATION
The clinical presentation of patients with aggressive NHL varies substantially based on histologic type and anatomic site of disease. The likelihood of B symptoms, including fever greater than 38°C, night sweats, or weight loss greater than 10% of body weight in the preceding 6 months, increases with NHL aggressiveness. Approximately 50% of patients present with these B symptoms, often with fatigue, malaise, and pruritus, although these are less common initially.
Most patients present with painless lymphadenopathy, which is often first treated with antibiotics for presumed infection and eventually biopsied when lymph nodes fail to regress. The most common scenario involves a diagnosis based on examination of peripheral lymph nodes, which may be detectable prior to internal lymph nodes becoming enlarged and causing symptoms. Peripheral lymph nodes are not usually painful, unless they are rapidly enlarging or are massive. Symptoms vary with the anatomic site of internal lymphadenopathy. Patients with mediastinal lymphadenopathy frequently experience cough, chest pain, and less commonly superior vena cava syndrome. Patients with large nodal masses in the abdomen or retroperitoneum frequently experience pain, abdominal fullness, or early satiety. Retroperitoneal lymphadenopathy can cause back pain and discomfort.
Extranodal disease is common in patients with aggressive NHL. The most common extranodal sites are the intestines, tonsils, and skin, although the frequency of involvement of these sites varies across reports. Disease in the gastrointestinal (GI) tract can present with nonspecific symptoms, including obstruction, blood loss with subsequent anemia, or diarrhea. Other extranodal sites include liver, lung, testis, bones, CNS, and spleen; however, aggressive extranodal NHL may involve nearly any organ system.
CLINICOPATHOLOGIC CHARACTERISTICS
Diffuse large B-cell lymphoma is the most common type of NHL (1,18). It occurs mainly in adults, with a median age in the sixth decade. Men are affected slightly more often than women. B-type symptoms or bulky disease occurs in one-third of patients. Nodal presentation is most common, but extranodal sites are involved in approximately 40% of patients (Figs. 8-1 and 8-2), and more than one-third of patients have more than one extranodal site of disease. Slightly more than half of patients have stage III or IV disease (19). Bone marrow involvement occurs in approximately 10% to 20% of patients. Diffuse large B-cell lymphoma uncommonly involves privileged sites, such as the testis and CNS, of which the latter portends a poor prognosis.
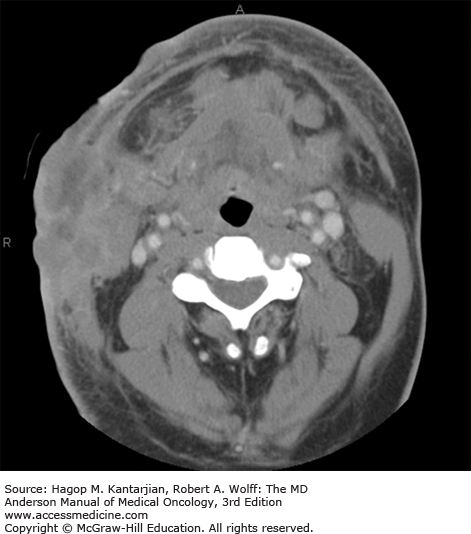
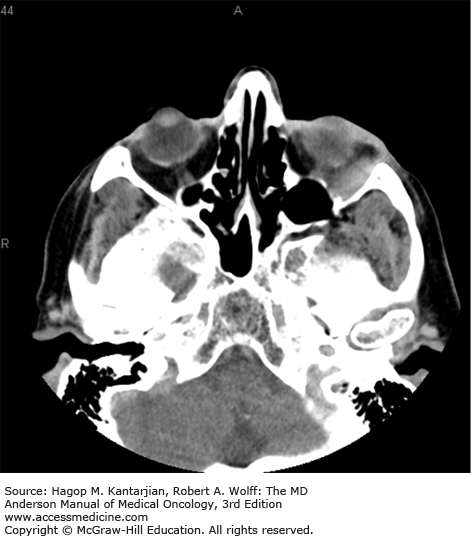
If untreated, DLBCL is invariably fatal, but DLBCL is generally sensitive to chemotherapy, at least initially. With current immune-chemotherapy approaches, a slight majority of patients will be cured of their disease, with 2-year event-free survival of approximately 70% (20). The International Prognostic Index (IPI) score remains a widely used prognostic model, despite not accounting for any tumor-specific biologic features. With rituximab plus chemotherapy–based disease management, the 4-year progression-free and overall survival rates for DLBCL patients with a Revised IPI (R-IPI) of 0, 1 to 2, and 3 to 5 are as shown in Table 8-2 (21).
These data are generally useful but unfortunately are not predictive of outcomes for an individual patient. Nearly half of patients will have a poor-risk R-IPI, and of these patients, approximately half will be cured and half will die of disease. Few patients with relapsed disease are cured.
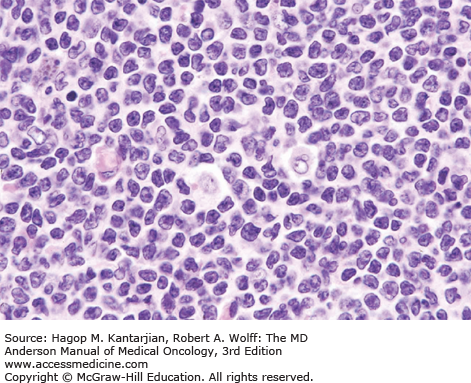
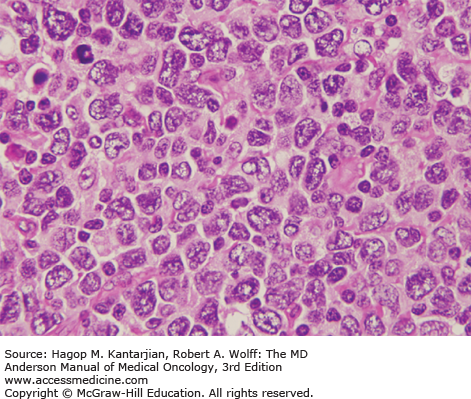
The diagnosis of DLBCL is based on its diffuse growth pattern and large neoplastic cells, with frequent mitotic figures (1). Cytologically, the neoplastic cells can be subdivided as large centrocytes, centroblasts, or immunoblasts. Large centrocytes range from 13 to 30 mm in size and have irregular or cleaved nuclear contours, relatively small, indistinct nucleoli, and a thin rim of eosinophilic cytoplasm. Centroblasts are 20 to 30 mm in size and have round or oval vesicular nuclei with two or three nucleoli and more abundant amphophilic cytoplasm (Figs. 8-3 and 8-4). Immunoblasts are larger than centroblasts, with an eccentrically located vesicular round or oval nucleus containing a prominent target-like central nucleolus and relatively abundant amphophilic, often plasmacytoid cytoplasm (Fig. 8-5). Elevated proliferation (Ki-67) rates and immunoblastic features appear to correlate with MYC rearrangements (22).
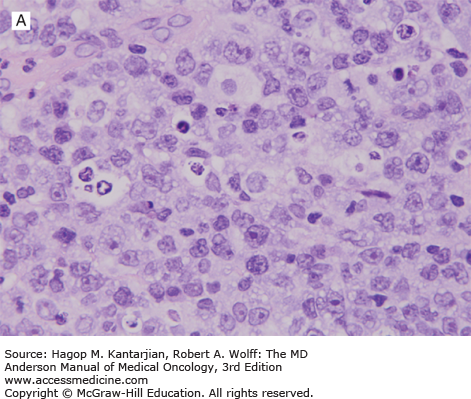
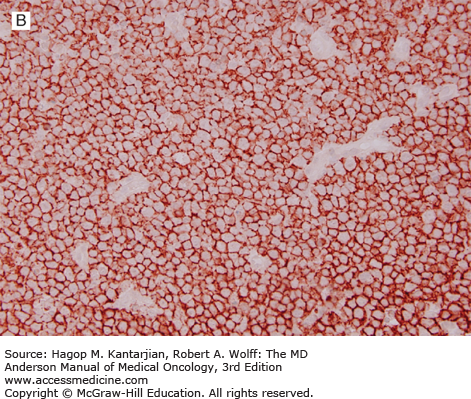
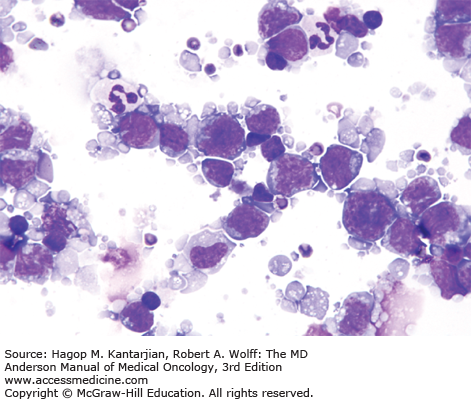
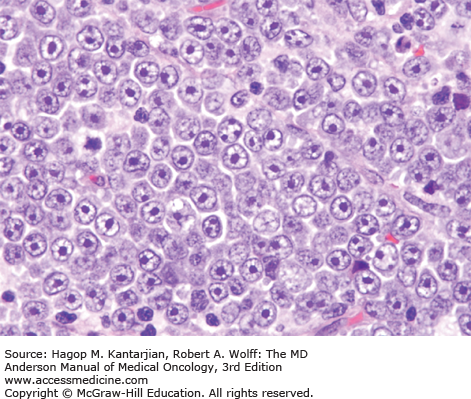
Immunophenotypic studies have shown that DLBCLs are of mature B-cell lineage. Approximately two-thirds of cases express monotypic immunoglobulin (Ig). These tumors express pan-B-cell antigens, 60% to 70% express BCL2, and a subset is positive for CD10 and BCL6.
Diffuse large B-cell lymphomas are heterogeneous at the molecular level. A subset of cases carries the t(14;18) involving BCL2, as shown by conventional cytogenetic or molecular studies (23,24). Another subset of DLBCLs has translocations or other abnormalities involving BCL6 at chromosome 3q27. BCL6 is rearranged in approximately 20% to 40% of DLBCLs, more often in tumors arising at extranodal sites (25). Recently, DLBCLs with translocations involving both MYC and BCL2 or BCL6, representing approximately 5% to 10% of DLBCL, have been shown to have an extremely aggressive behavior and are commonly referred to as “double-hit” DLBCL (26). Diffuse large B-cell lymphomas with overexpression of MYC and BCL2 or BCL6 protein levels are more common and are designated double-positive DLBCLs. These tumors are still adverse compared to DLBCLs without protein overexpression but less so than translocation-defined double-hit DLBCLs (27,28,29).
Gene expression profiling (GEP) can molecularly divide DLBCL into three groups: germinal center B-cell like (GCB), activated B-cell like (ABC), and a third noncharacteristic group. Patients with the GCB type of DLBCL have a better prognosis independent of the IPI (30,31). Despite the initial GEP studies of DLBCL occurring approximately 15 years ago, this technology is not routinely used in clinical practice due to logistical issues and need for significant bioinformatic analysis. Fortunately, new technologies may allow these issues to finally be overcome by limiting the number of genes analyzed, such as in the Lymph-2Cx Nanostring assay (Nanostring, Seattle, WA) (32). In lieu of GEP, a limited number of immunohistochemical markers, including CD10, MUM-1, BCL6, GCET1, and FOXP1, can subclassify DLBCL into GCB and non-GCB with relatively good concordance with GEP (33,34).
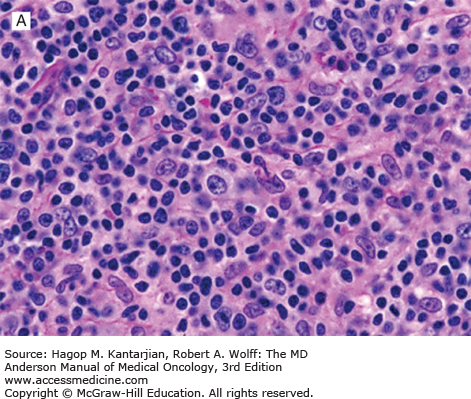
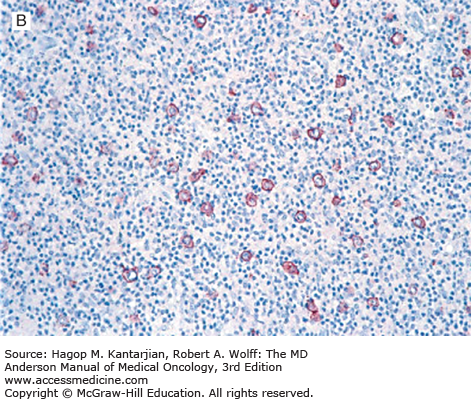
T-cell/histiocyte-rich (TCHR) DLBCL is a diffuse neoplasm in which most of the cells are reactive T cells and histiocytes and the large, neoplastic B cells represent <10% of all cells in the infiltrate (Fig. 8-6). Patients with TCHR-DLBCL commonly have a history of nodular lymphocyte-predominant Hodgkin lymphoma, and TCHR-DLBCL may represent a transformation event in some patients.
FIGURE 8-6
T-cell/histiocyte-rich large B-cell lymphoma. A. Scattered large neoplastic lymphoid cells in a background of numerous small lymphocytes. B. The large neoplastic cells are positive for CD20, and the small lymphocytes are T cells (immunostain not shown [A, hematoxylin-eosin, 630×]; B, immunohistochemistry, 200×).
This entity includes all primary intracerebral or intraocular lymphomas. Although these lymphomas are remarkable for their unique clinical presentation, histologically and at the immunophenotypic level, these neoplasms closely resemble other cases of systemic DLBCL. In HIV-positive patients, the neoplastic cells commonly have immunoblastic features and are positive for EBV. Approximately, one-third of the cases demonstrate translocations involving the BCL6 gene.
As the name suggests, DLBCL, leg type, presents as cutaneous lesions originally described in the lower extremities, although all skin sites may be affected. Lesions often are initially confused with an insect bite, but eventually become diffuse and ulcerated, and the clinical course is aggressive. Histologic sections show diffuse sheets of monotonous neoplastic cells with centroblastic or immunoblastic morphology with frequent mitotic figures (36). The neoplastic cells have a non–germinal center B-cell immunophenotype, and fluorescence in situ hybridization studies often detect rearrangements of BCL6, IGH, and/or MYC (37).
This DLBCL is most common in the elderly, defined arbitrarily as >50 years of age but can occur in younger patients and often presents at an advanced stage. Epstein-Barr virus is a defining feature. Histologically, these neoplasms have a diffuse pattern and show a spectrum of features, and two morphologic variants are recognized: polymorphous, with a broad range of B-cell maturation in the reactive background, and monomorphous, which contains mostly large cells (1). Geographic necrosis is common, and the neoplastic cells usually have a non–germinal center B-cell immunophenotype.
These are rare neoplasms in which the lymphoma cells are confined to intravascular spaces (1). The histologic diagnosis can be subtle, and therefore, diagnosis can be delayed. The neoplastic cells are large, express B-cell antigens, and usually have a non–germinal center B-cell immunophenotype. The molecular basis of intravascular large B-cell lymphoma is unknown (Fig. 8-7).
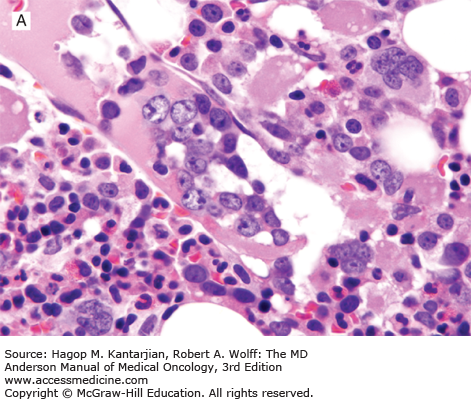
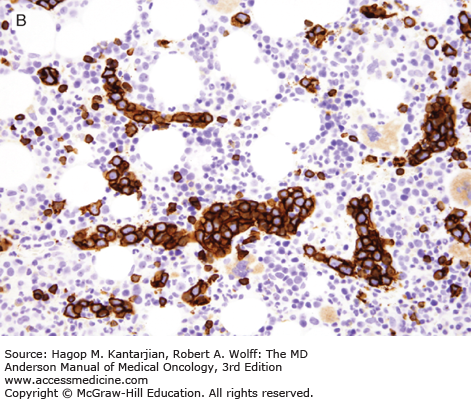
Primary effusion lymphoma, also known as body cavity–based lymphoma, is a very rare neoplasm of large B cells that involves a body cavity and is almost always associated with AIDS (38). The prognosis for patients with PEL is poor. Human herpesvirus type 8 (also known as Kaposi sarcoma–associated herpesvirus) is present in virtually all cases of PEL, and its presence selects for a distinct cellular gene expression profile (1). EBV is also present in most cases of PEL. An extracavitary (solid) variant of PEL also can occur, involving lymph nodes or extranodal sites (39).
These neoplasms are thought to arise in the thymus, are usually localized to the thoracic cavity, and occur more frequently in young women, with a female-to-male ratio of 2:1 (1). Patients with primary mediastinal B-cell lymphoma (PMBL) present frequently with cough and dyspnea mimicking a respiratory infection or, rarely, superior vena cava syndrome. Histologically, PMBL has a diffuse pattern and is composed of large lymphoid cells that exhibit a spectrum of cytologic appearances: centroblastic, immunoblastic, or a mixture. Sclerosis is common, mitotic figures are usually numerous, and the tumor cells can have clear or pale cytoplasm. Immunophenotypic studies have shown that PMBLs are frequently Ig negative and commonly lack major histocompatibility complex class II antigens. Gene expression profiling analysis of PMBL has shown significant overlap with classical Hodgkin lymphoma (40). Rearrangements of CIITA occur in about 40% of PMBLs, and amplification of the chromosome 9p24.1 locus (site of programmed death ligands) and mutations of PTPN1 have been reported in about 25% of cases (41). These molecular abnormalities highlight the interaction between the immune system and PMBL in pathogenesis and suggest potential therapeutic targets.
Mantle cell lymphoma (MCL) represents approximately 6% of all NHLs (19). Patients with MCL have a median age in the seventh decade, and MCL has a male-to-female ratio of approximately 3:1 (42). Most patients present with advanced-stage disease, with bone marrow involvement in approximately 60% of patients, and most patients have low-level involvement of the peripheral blood. Overt leukemia may be associated with a poorer prognosis. Although GI symptoms are uncommon, 85% to 90% of MCL patients have GI involvement (43). Blastoid MCLs have a more aggressive clinical course and often have occult CNS involvement. In the peripheral blood, MCL can sometimes resemble B-cell prolymphocytic leukemia.
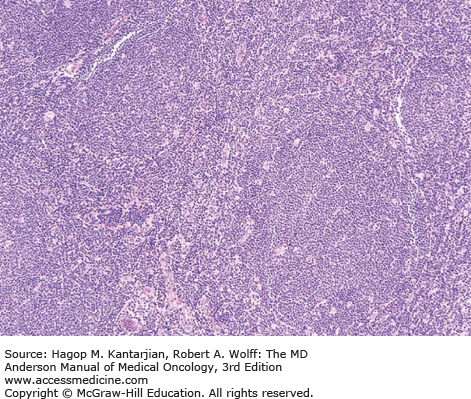
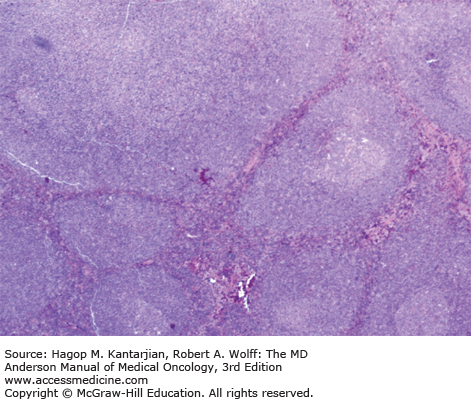
Histologically, in classical MCL, the lymph node architecture is replaced by a diffuse or vaguely nodular neoplasm (Fig. 8-8) (1). In a subset of cases, a mantle zone pattern results when the neoplasm selectively involves the follicular mantle zones surrounding normal or reactive germinal centers (Fig. 8-9). Cytologically, MCL is composed of a monotonous population of small lymphoid cells with slightly or clearly irregular nuclear contours (Fig. 8-10). In about 15% of cases, MCL may show blastoid (lymphoblastic-like) or pleomorphic (large cell–like) features. These neoplasms are associated with a poorer prognosis and have a high frequency of TP53 or TP16 mutations.
Mantle cell lymphomas express monotypic Ig light chain IgM, IgD, pan-B-cell antigens, BCL2, SOX11, alkaline phosphatase, and CD5 (21). A high proliferation rate, most often assessed by Ki-67 immunohistochemical analysis, predicts a poorer prognosis. t(11;14)(q13;q32) is present in virtually all cases of MCL (44). In this translocation, CCND1 on 11q13 is juxtaposed with IGH on 14q32, resulting in overexpression of cyclin D1 (Fig. 8-11). Cyclin D1 facilitates cell cycle transition from G1 to S phase (45). Although t(11;14) is central to the pathogenesis of MCL, t(11;14) is insufficient to cause lymphomagenesis. Conventional cytogenetic studies have shown a number of additional abnormalities (46,47), and a number of gene mutations have been reported.
There are three variants of BL: endemic (African), sporadic (nonendemic), and AIDS-associated (21,48). Endemic BL was first described in equatorial Africa and is associated with EBV infection in 95% of patients (48). The median age of patients is approximately 7 years, with a 3:1 male-to-female ratio (49). Abdominal and retroperitoneal lymph nodes, the GI tract, and the gonads can be involved.
Sporadic BL occurs in industrialized nations and is associated with EBV infection in approximately 25% of patients. Patients are usually in the second or third decade of life, with a male-to-female ratio of 3:1. Involvement of the jaw is uncommon, and patients may present with large masses in the abdomen, peripheral lymph nodes, pleura, or pharynx (19). In endemic or sporadic BL, bone marrow and CNS involvement are uncommon at presentation, but they are frequent sites of subsequent dissemination.
Burkitt lymphoma can also occur in the clinical setting of immunosuppression, including HIV infection, posttransplant, or congenital immune deficiency setting (19). Epstein-Barr virus infection occurs in 30% to 40% of cases.
Morphologically, all variants are similar. The relatively clear histiocytes in a background of blue lymphoma cells imparts a “starry sky” appearance (Fig. 8-12A). This pattern results from rapid cell turnover with individual cell necrosis and scavenging of debris by macrophages. The neoplastic cells are round to ovoid, uniform in shape, and approximately the size of benign histiocyte nuclei. The chromatin is coarse, with two to five prominent basophilic nucleoli. Mitotic figures are numerous.
FIGURE 8-12
Burkitt lymphoma. A. The neoplastic cells are intermediate in size, similar to that of benign histiocyte nuclei, with multiple small nucleoli. A starry sky pattern is also seen in this field. B and C. The neoplastic cells are negative for BCL-2 (B) and are >99% positive for Ki-67 (C). (A, hematoxylin-eosin, 1,000×; B, C, immunohistochemistry, 400×.)
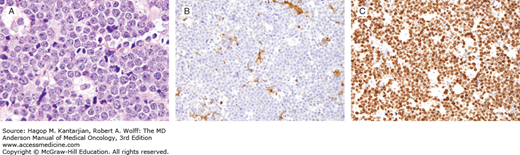
Burkitt lymphomas of endemic, sporadic, and AIDS-associated types are of mature B-cell lineage and express Ig, pan-B-cell antigens, CD10, and BCL6 (23). Burkitt lymphomas have a very high proliferation rate (>99%) using an antibody specific for Ki-67 (Fig. 8-12B and C) and are negative for BCL2. MYC translocations are characteristic of BL. Approximately, 80% of cases carry the t(8;14)(q24;q32), with the remaining cases having one of two variant translocations, t(2;8)(p11;q24) or t(8;22) (q24;q11) (50,51,52). Common to each of these translocations is involvement of chromosome region 8q24, the site of the MYC, which is deregulated. Via these translocations, MYC is juxtaposed with the IgH or the Ig light chain genes.
B-Cell Lymphoma With Features Intermediate Between Diffuse Large B-Cell Lymphoma and Burkitt Lymphoma
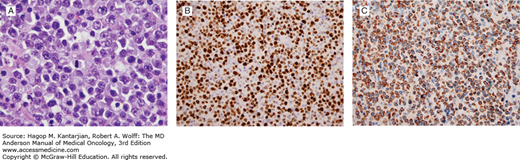
This entity in the 2008 edition of the World Health Organization (WHO) classification of hematopoietic neoplasms is designed for cases that do not fit either DLBCL or BL (1). In the past, these cases were designated as atypical Burkitt/Burkitt-like lymphoma or as high-grade B-cell lymphoma or B-cell lymphoma with high-grade features. This group often exhibits morphologic and immunophenotypic deviation from typical cases of BL and DLBCL. These neoplasms have a diffuse pattern, often with a starry sky appearance, and a high proliferation rate (Fig. 8-13) (53,54). A significant number of cases demonstrate MYC rearrangement, although in some of the cases, MYC rearrangement involves one of the non-Ig partners (55). Many cases of so-called double-hit lymphoma fit within this category. These cases commonly demonstrate strong BCL2 expression and a complex karyotype (56). The algorithm for diagnosis of high-grade B-cell lymphoma is illustrated in Fig. 8-14.
FIGURE 8-13
B-cell lymphomas with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma. Similar to Burkitt lymphoma, the neoplastic cells are intermediate-sized and demonstrate brisk mitotic activity, but unlike Burkitt lymphoma, cells have prominent single nucleoli (A). While the proliferative rate is similar to Burkitt lymphoma (B), the neoplastic cells strongly express Bcl-2 (C). In this case, fluorescence in situ hybridization detected both translocations t(8;14) and t(14;18), so-called double-hit lymphoma (Lymph node, A, hematoxylin-eosin, 1,000×; B, Ki-67 (Mib-1), 400×; C, Bcl-2, 400×).
STAGING AND INITIAL EVALUATION
The Ann Arbor Staging system, developed in 1971 for Hodgkin lymphoma, is used to stage NHL (Table 8-3) (19). NHLs are often disseminated at diagnosis, unlike Hodgkin lymphoma. Of interest, there does not appear to be any meaningful difference in the outcomes of patients with stage III or IV disease, and thus the purpose of staging is to identify the patients with localized NHL who may benefit from additional local therapy.
| Stage I | Involvement of a single nodal group or extranodal site (IE) |
| Stage II | Involvement of two or more nodal groups on the same side of the diaphragm or localized involvement of an extranodal site or organ (IIE) and one or more nodal groups on the same side of the diaphragm |
| Stage III | Involvement of nodal groups on both sides of the diaphragm, which may be accompanied by localized involvement of an extranodal region or site (IIIE) of spleen (IIIS) or both (IIISE) |
| Stage IV | Diffuse or disseminated involvement of one or more distant extranodal sites |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


