 FIGURE 54.1 Schematic representation of functional regions of fibrinogen and fibrin. The C-terminal sequences of the γA and γ′ chains are amplified for clarity, showing the high affinity thrombin binding, platelet interaction, and factor XIIIa catalyzed cross-linking (XL) sites. Elsewhere on the D region are the ‘a’ and ‘b’ pockets, and the D:D interface indicating the sites between D s of two molecules that associate during fibril elongation. The lower left panel indicates the slight weakening of αC tethering to the E region by FpA release, reflecting the contributing role to the tether by intact FpA.131 The lower right panel illustrates the untethering and dissociation of αC s following FpB release.128,132 |
fragment D-D in the presence of peptides GlyProArgPro-amine (GPRPam) and GlyHisArgPro-amine (GHRPam), which mimic the N-terminal α and β sites of fibrin, respectively.54,55,56,57 Substitutions occurring within these crystal structures have enabled correlations of clinical and biochemical data with the three-dimensional fibrinogen structure. Mutations in the E region (Table 54.2) impair one or more of the following functions: thrombin binding to fibrinogen, fibrinopeptide cleavage, and fibrin polymerization.
Table 54.1 Mutations resulting in afibrinogenemia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
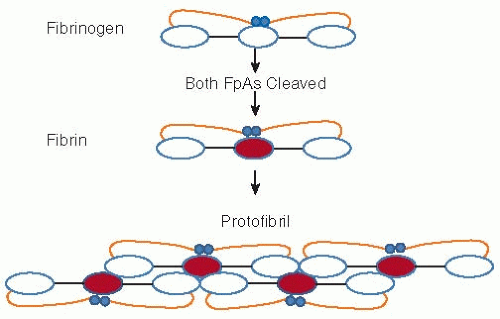 FIGURE 54.2 Schematic representation of fibrinogen and its conversion to fibrin. Release of FpA initiates polymerization. Each central (or E shown in red) fibrin region associates with the outer (or D) regions of two other molecules initiating a double stranded, half staggered, two molecule thick fibril, known as protofibril. As elongation progresses FpB release, significantly slower than that of FpA, is accelerated. Following FpA release, illustrated in Fig. 54.1, the αCs become untethered and dissociate. Each free αC associates with one or more counterpart(s) on neighboring fibrils (not shown) and in this manner promotes lateral assembly. |
Unexpectedly, the heterozygous AαV20D substitution59 results in absence of residues Aα1-20, thus explaining the approximately half of normal amount of released FpA. This apparently reflects a new furin-susceptible Aα20-21 bond, which enables intracellular removal of the mutant peptide.
Table 54.2 Mutations resulting in hypofibrinogenemia | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
in the C-terminal region in the β chain (β397-432) of another fibrin(ogen) molecule. This interaction is modeled in the crystal structures obtained in the presence of GHRPam (see previous reviews7,8). Only one substitution has been found in the “B” knob: heterophenotypic BβG15C (Ise, Fukuoka II, Kosai, Ogasa, and Hamamatsu II). Functional studies showed normal FpA release, approximately 50% FpB release, impaired thrombin- and reptilase-catalyzed polymerization, and impaired desA- and desAB-fibrin monomer repolymerization.73,74 In addition to impairing the function of “B” knob, these results suggest they impair that of the “A” knob whose second part8 resides within β.15-42 The sulfhydryl group of each varied. Some formed complexes with albumin attributable to disulfide links via the albumin’s solitary-free C34 sulfhydryl,75 some formed fibrinogen-fibrinogen dimers, and some were free sulfhydryls. Three were asymptomatic and two were associated with thrombosis.
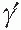 chain is impaired.81 The second is a heterophenotypic fibrinogen (New York I) with a large deletion of residues Bβ9-72, encoded by FGB exon 2.82 Its delayed FpA and FpB release is also attributable to diminished thrombin binding. A third substitution, a heterophenotypic BβA61K (Miami),83 is associated with repeated spontaneous abortions and impaired polymerization. A reptilase-incoagulable subfraction (60% of plasma fibrinogen) displayed thrombin binding <10% of normal controls (author’s unpublished data).
chain is impaired.81 The second is a heterophenotypic fibrinogen (New York I) with a large deletion of residues Bβ9-72, encoded by FGB exon 2.82 Its delayed FpA and FpB release is also attributable to diminished thrombin binding. A third substitution, a heterophenotypic BβA61K (Miami),83 is associated with repeated spontaneous abortions and impaired polymerization. A reptilase-incoagulable subfraction (60% of plasma fibrinogen) displayed thrombin binding <10% of normal controls (author’s unpublished data).Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Blood Coagulation Factors and Inhibitors: Their Primary Structure, Complementary DNAs, Genes, and Expression
Fibrinogen Structure and Function
Megakaryocyte Structure and Platelet Biogenesis
Integrín αIIbβ3 and Platelet Aggregation
Nitric Oxide: The Vascular Biology of Nitric Oxide and Nitric Oxide Synthases
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Blood Coagulation Factors and Inhibitors: Their Primary Structure, Complementary DNAs, Genes, and Expression
Fibrinogen Structure and Function
Megakaryocyte Structure and Platelet Biogenesis
Integrín αIIbβ3 and Platelet Aggregation
Nitric Oxide: The Vascular Biology of Nitric Oxide and Nitric Oxide Synthases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


