While the overall prevalence of adrenal tumors is estimated to be 3% in those >50 years of age and 6% in those >60 years of age, primary malignancies of the adrenal glands are extremely rare.14,51,52 When discovered during imaging for reasons other than an evaluation for suspected adrenal diseases or staging for a different primary tumor, adrenal tumors are referred to as adrenal incidentalomas. The vast majority of adrenal incidentalomas are benign, but up to 15% can be functional, autonomously secreting adrenal hormones leading to clinical or subclinical hormone excess, making clinical exam and biochemical evaluation for hormone excess mandatory.3,6,7,14
The estimated incidence of ACC is 0.5 to 2 per million per year. It has a slight predilection for women (female:male, 1.5:1) with a relative bimodal age distribution peaking in early childhood and in the fifth decade of life.3,53,54–56 Although there are no established risk factors for sporadic ACC, smoking and early contraceptive use have been suggested.57,58 Up to 10% to 15% of all ACCs arise in patients with familial cancer syndromes, such as Li-Fraumeni syndrome, Lynch syndrome, multiple endocrine neoplasia type 1, and less commonly familial adenomatous polyposis, Beckwith-Wiedemann syndrome, or Carney complex.59,60
The overall rate of PCPGL in the general population has been estimated at between 0.05% to 0.1%.61,62 The estimated incidence of malignant PCPGL is 1 to 5 per million per year.26 Between 3% to 36% of PCPGL are malignant, with higher rates of malignancy in children, those with SDHB mutations, and those with extra-adrenal tumors.26,29,62,63 The average age at presentation is 44 in sporadic cases, compared with 25 for hereditary disease.62,64,65 Although historically only 10% of PCs were assumed to be associated with a familial syndrome, this is now estimated at 25% to 30%, particularly when also including PGLs.26,63 The relatively high contribution of hereditary conditions predisposing to all adrenal neoplasms warrants evaluation of every patient by a cancer geneticist and genetic counselor.
SCREENING IN SPECIAL POPULATIONS
There are no current screening recommendations for adrenal tumors in the general population. As such, screening should only be considered for patients with a known or suspected familial syndrome that predisposes to adrenal neoplasia. Of all familial syndromes associated with ACC, Li-Fraumeni syndrome has the highest lifetime risk at up to 6.5% to 9.9%.66,67 Adrenal lesions identified in imaging obtained in patients with any of the other predisposing syndromes should be evaluated carefully for signs of malignancy. All patients diagnosed with PCPGL should undergo evaluation for predisposing familial syndromes caused by mutations in SDHx, VHL, NF1, TMEM127, MAX, and RET. Demonstration of a genetic mutation allows for identification of other affected family members and disease-specific screening and surveillance for adrenal neoplasms and other associated tumors in all gene carriers.49,68,69
DIAGNOSIS OF ADRENAL NEOPLASIA
Patients with adrenal neoplasia may seek medical attention because of symptoms related to the size of their mass, hormone excess, or be referred for an incidentally discovered tumor (Fig. 84.1).64,70 Evaluation should begin with a thorough history and physical exam, including specific questions regarding symptoms of hormone excess, systemic illness, and a careful family history. The classic symptom triad of PC is headache, palpitations, and diaphoresis, but presentation is highly variable and the complete triad is not observed in all cases.26,64 Patients with functional cortical neoplasms may present with overt hypercortisolism, hyperaldosteronism, virilization, or rarely feminization. Cortisol is the most commonly produced hormone in both benign and malignant functional cortical neoplasms.14,15,70 Mild hypercortisolism may be challenging to recognize given the absence of obvious signs.6,7 Hypertension may be caused by aldosterone-producing ACA, although aldosterone secretion by ACC is rare. Hypertension in ACC is more often related to mineralocortioid effects of cortisol or secreted steroid intermediates.14,70 Virilization or feminization should increase suspicion for malignancy, as adrenal androgens are the next most common steroid hormone secreted by ACC, and up to half of hormone-producing ACCs produce both cortisol and androgen.70 Likewise, hypercortisolism of rapid onset should increase concern for ACC.
Basic laboratory evaluation of a patient presenting with an adrenal mass or syndrome of hormone excess includes a complete blood count and comprehensive metabolic panel with liver enzymes. Those presenting with an incidental adrenal mass require comprehensive hormonal evaluation including tests for hypercortisolism, androgen excess, hyperaldosteronism in hypertensive patients, and catecholamine production in all patients.71,72 The most sensitive test for hypercortisolism is a 1 mg dexamethasone suppression test. Overt hypercortisolism is most often associated with morning (8 a.m.) cortisol levels >5 μg/dL following dexamethasone testing, while a value of <1.8 μg/dL excludes endogenous hypercortisolism.7 Conversely, spontaneous morning (8 a.m.) ACTH levels of <10 pg/ml will be demonstrated for most cortisol-secreting tumors due to suppression of the hypothalamic-pituitary axis (HPA).6 Bioavailable or total testosterone and dehydroepiandrosterone sulfate are obtained to evaluate potential androgen excess, the latter produced only in the adrenal gland and therefore helpful in differentiating adrenal from gonadal neoplasia. Hypertensive or hypokalemic patients should have serum aldosterone concentration and plasma renin activity (PRA) measured. Hyperaldosteronism is typically diagnosed with a ratio of serum aldosterone concentration/PRA >20 to 40, provided the absolute level of aldosterone is also elevated, since mineralocorticoid effects of intermediates or volume repletion may also suppress renin.7 Plasma free metanephrine and normetanephrine are the most sensitive tests for PCPGL.73 Positive screening hormonal evaluation dictates additional confirmatory hormonal studies, which may include 24-hour urine free cortisol to measure the extent of hypercortisolism,6 serum aldosterone levels following saline-infusion or 24-hour urine aldosterone under salt loading conditions, or measurement of additional steroid hormones such as progesterone, androstenedione, estradiol, 17-hydroxyprogesterone, or 17-hydroxypregnenolone for further characterization.6,7,72 The initial steroid profile can be useful for surveillance in ACC.21,72 For PCPGL, chromogranin A should be obtained, as it can be followed in surveillance.26,74–76
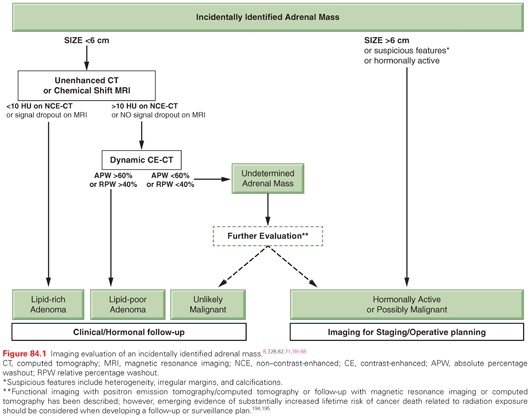
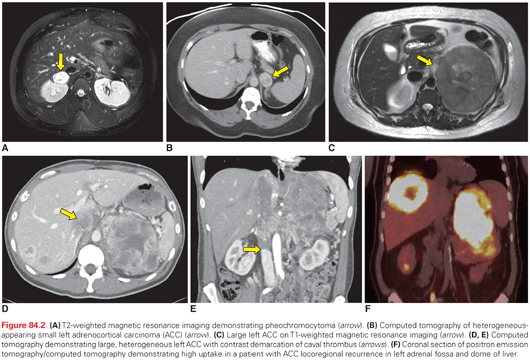
Cross-sectional imaging (Fig. 84.2) with computed tomography (CT) or magnetic resonance imaging (MRI) is important for establishing the extent of disease and identifying features concerning for malignant cortical lesions. Benign cortical lesions tend to be small, fat-containing, and homogeneous, with smooth borders. Conversely, large, heterogeneous lesions with irregular margins and calcifications are strongly suggestive of ACC. The risk of ACC increases with size, and is estimated at <2% for lesions <4 cm, 2% to 6% for lesions 4 to 6 cm, and 25% for lesions >6 cm. Lesions with enhancement of <10 Hounsfield units (HU) are indicative of lipid-rich ACA, although up to 30% of ACAs may be lipid-poor with intermediate enhancement between 10 and 30 HU.6,7,62,77 Lipid-containing lesions may also be identified on MRI by signal dropout on opposed-phase sequences. PCPGL are often bright as cerebrospinal fluid on T2-weighted MRI, but can be variable in appearance. They tend to be homogeneous when smaller and heterogeneous when larger with features overlapping lipid-poor ACAs, but no imaging features predict malignancy.7,26,62
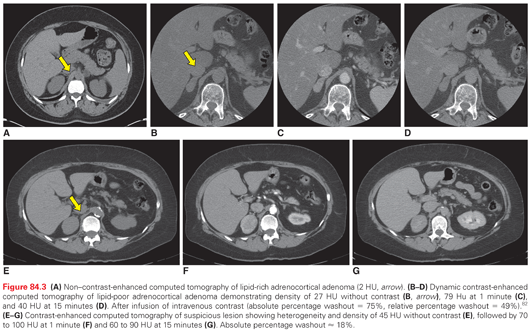
Suspicious tumors or those with indeterminate characteristics require additional imaging to further characterize the lesion in question and to evaluate for metastatic disease.78 If the initial CT was non–contrast enhanced or optimized for another purpose, a dedicated adrenal-protocol CT with thin slices, oral contrast, and careful timing of intravenous contrast should be obtained for lesions that are homogeneous on the initial scan.62 Absolute percentage enhancement washout above a threshold of 60% (or relative washout above 40%) at 15 minutes has high sensitivity and specificity for lipid-poor ACA (Fig. 84.3).79–82 Compared with CT, contrast-enhanced MRI is more sensitive for vascular invasion and is occasionally obtained as a complementary study in patients with larger tumors.71 Contrast-enhanced CT of the chest, abdomen, and pelvis is appropriate to evaluate for metastases and hence complete staging.71 Imaging of the head should only be obtained if neurologic symptoms are present, and bone scintigraphy can be obtained for those presenting with skeletal pain. Neither test is required in the initial evaluation of the asymptomatic patient.
Functional imaging can be useful in evaluating both cortical and medullary tumors of the adrenal gland. Deoxy-2[18F]fluoro-d-glucose positron emission tomography (18FDG-PET) (Fig. 84.2F) is a useful, complementary modality that can sometimes alter management of an indeterminate lesion (i.e., one that is unable to be characterized with a high degree of certainty as benign or malignant). However, no validated quantitative criteria exist for reliably predicting whether a lesion that is indeterminate by anatomic imaging in a patient without a known primary cancer will be benign or malignant.78,83–88 Furthermore, metastatic disease from nonadrenal primary, PC (benign or malignant), and up to 10% of ACA can be positive on 18FDG-PET scan.79,89–93 123I-metaiodobenzylguanidine (123I-MIBG) scans have high positive-predictive value for PCPGL and can be used for the identification of additional foci of disease, including multicentric tumors or metastasis, especially in patients with inherited syndromes.26,94 However, the use of 123I-MIBG for diagnostic purposes is of rather limited utility as the diagnosis of PCPGL is often made through simple biochemistry and cross-sectional imaging. Moreover, for PGL of the head and neck 111In-pentetreotide scintigraphy has better sensitivity.95 It is worth mentioning that functional imaging will rarely discover any tumor that is not evident on cross-sectional imaging. In such cases where biochemical testing suggests PCPGL but imaging is negative, there are often other causes for clinical symptoms and/or elevated metanephrines (e.g., use of tricyclic antidepressants, congestive heart failure).
Percutaneous biopsy of adrenal tumors is almost never indicated because cytology is unhelpful in characterizing adrenal neoplasia, and biopsy has significant risk of complications including hemorrhage and pneumothorax.6,7,96–98 Careful consideration should be given to how the test will influence management and whether alternative approaches, such as PET/CT, can suffice, as most single adrenal masses can be diagnosed by imaging and endocrine evaluation.71,89 Only in the setting of a patient with a nonadrenal primary cancer should an adrenal biopsy be considered if it remains unclear whether the adrenal mass is a primary adrenal tumor versus a metastasis of the nonadrenal primary tumor. PCPGL should always be excluded prior to an attempted biopsy due to the high morbidity and mortality of biopsy-associated catecholamine surges.97,98
GRADING AND STAGING OF MALIGNANCY
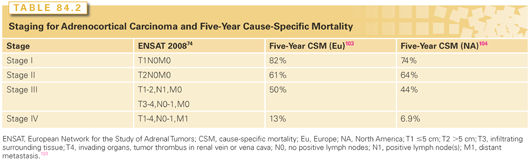
Establishing malignancy of adrenal neoplasms can be challenging. Histopathologic criteria established by Weiss predict malignancy in adrenocortical neoplasms when three or more of nine criteria are present.71,99 Mitotic rate is a particularly important criterion and indicates malignancy when greater than five mitoses are observed in 50 high-powered fields. Mitotic activity >20/50 high-powered fields defines a high-grade ACC.71,100,101 Several staging systems have been proposed for ACC, but the European Network for the Study of Adrenal Tumors staging system (Table 84.2) has become widely adopted as it best reflects outcome.102–105
Differentiation between benign and malignant PCPGL is a challenging clinical problem as well. Indeed, because even extensive invasion does not accurately predict malignancy, the only criterion adopted by the World Health Organization for defining malignant PCPGL is metastasis.73 No single histologic criterion predicts malignant PCPGL. The Pheochromocytoma of the Adrenal Gland Scaled Score system has been proposed for assessing malignant potential by using 12 histopathologic criteria,106 but validation studies have produced conflicting results.107–109 At present, there is no staging system for PCPGL. Tumors are categorized as local, regional, or metastatic.62
IMPORTANT CONSIDERATIONS IN PERIOPERATIVE MANAGEMENT OF ADRENAL TUMORS
Patients with adrenal neoplasms present unique perioperative challenges. In addition to hemodynamic and metabolic instability due to intraoperative catecholamine fluctuation, patients with PCPGL may have established cardiovascular sequelae related to chronic catecholamine excess.110 Those with cortisol-secreting tumors are at higher risk for infectious, metabolic, and wound-healing complications, and will require careful glucocorticoid replacement following complete surgical resection due to suppression of the HPA.70
Historically, perioperative mortality related to resection of PCPGL was as high as 45%. The introduction of α-blockade and volume repletion has reduced mortality to 0% to 3% in contemporary series.69,111 It is important to recognize that even patients with asymptomatic PCPGL are at risk for cardiovascular complications without preoperative α-blockade and that significant elevations in blood pressure may still occur intraoperatively despite adequate α-blockade.29,112 Either selective or nonselective α antagonists can be used to establish α-blockade. Selective α1-blockade has been used to avoid some undesirable α2-mediated side effects including reflex tachycardia, hypoglycemia, and somnolence, but these agents are less well studied compared with nonselective agents. Traditionally, phenoxybenzamine is the preferred nonselective α-blocker for PCPGL. Dosage can be titrated on an outpatient basis for adequate blockade, which often takes 2 weeks or longer. During this time, salt- and fluid-loading are recommended for volume optimization to minimize postoperative hypotension.69 Other α-blocking agents can serve as an alternative to phenoxybenzamine and are preferred by some institutions.113 Initiation of a β-blocker for reflex tachycardia may be considered, but not before α-blockade is established. Data regarding the use of calcium channel blockers and angiotensin-converting enzyme inhibitors preoperatively in patients with PC are limited.114–116 It is important to recognize that α-adrenergic vasopressors may not be helpful in treating hypotension resulting from α-blockade and that volume expansion should be the treatment of choice.69
Resection of cortisol-producing tumors may present unique perioperative challenges as well. Aggressive preoperative control of hypercortisolism is advocated but should not delay surgery.70 Postoperative adrenal insufficiency should be anticipated in all patients with preoperative hypercortisolism and replacement should be initiated preemptively postoperatively, with surveillance of the HPA and treatment adjustment as necessary. Recovery of the HPA might take up to 6 to 24 months, and additional steroid replacement during times of stress to prevent Addisonian crisis is necessary.71,117,118
MANAGEMENT OF BENIGN-APPEARING ADRENOCORTICAL TUMORS AND PHEOCHROMOCYTOMA
Before pursuing surgical management, an assessment of the likelihood of malignancy based on size, imaging, and clinical presentation must be made.119 The surgeon should strictly adhere to oncologic principles when resecting tumors not meeting criteria for being benign.70
Although its role in potentially malignant lesions is controversial, minimally invasive adrenalectomy has lower morbidity, decreased pain, and shorter hospital stay, and is the procedure of choice for benign-appearing adrenocortical tumors or PCPGL if technically feasible.120–122 Minimally invasive approaches for adrenalectomy may be performed laparoscopically by an anterior abdominal approach or by a posterior retroperitoneal approach.120,123 The posterior retroperitoneal approach is advantageous in patients with prior abdominal operations because the potential need for adhesiolysis is obviated; however, the posterior retroperitoneal approach is not advised in patients with significant subcutaneous or perinephric fat, unfavorable anatomy, or tumors >8 cm.124,125 Partial (i.e., cortical-sparing) adrenalectomy is the preferred approach for patients with familial bilateral benign diseases, such as bilateral PCPGL in patients with von Hippel-Lindau disease or multiple endocrine neoplasia type 2.29,126,127
SURVEILLANCE AFTER RESECTION OF PHEOCHROMOCYTOMA
There are no standard recommendations for surveillance for malignant PCPGL after resection of apparently benign disease. However, most endocrine neoplasia groups suggest that all patients receive some form of long-term follow-up.29,69 Surveillance strategies must be individualized and may depend on overall impression of risk for malignancy (e.g., SDHB), operative findings, and genetic predisposition to additional primary tumors.128 Initially, all patients should undergo surveillance reevaluation with history, physical exam, laboratories, and imaging. Postoperative biochemical testing at 2 to 6 weeks and 6 months is appropriate, and chromogranin A may be useful if elevated preoperatively.29,69,94 The preferred imaging modality for surveillance is CT or MRI of the site of the initial tumor, as local recurrence is the most common form of relapse. Particularly in patients with hereditary syndromes, MRI should be preferred as the number of scans might otherwise lead to excessive radiation doses.94
MANAGEMENT OF RECURRENT OR METASTATIC MALIGNANT PHEOCHROMOCYTOMA
Diagnosis of malignant PCPGL is made based on recurrence or identification of metastasis on initial presentation.129 In a retrospective review of 176 patients with PCPGL initially presenting without evidence of malignancy, up to 17% of patients developed recurrence at either the primary site or another location. Recurrences were malignant in half of all cases and more commonly occurred in patients with familial syndromes or larger tumors.130 The most common sites of metastasis of PCPGL are to bones, lungs, liver, and lymph nodes.69,94 Five-year survival is 34% to 60%, although this is dependent on location of metastasis, as those with lung and liver lesions have shorter survival compared with those who have osseous metastasis only.29
Treatment options for malignant PCPGL are limited and the goal of therapy is palliative, with a focus on controlling catecholamine secretion, pain, and tumor burden.72,131,132 Options include supportive medications, modalities for locoregional control, external-beam radiation, radiopharmaceuticals, and chemotherapy. Quality of life must be considered because in some cases observation and supportive medication alone is the best option.72,131 Debulking and metastasectomy can be considered in patients who have significant resectable tumor burden in the context of slowly progressive disease.26,72,131,132 Radiofrequency ablation (RFA) and transarterial chemoemoblization (TACE) have been described for unresectable metastases. Similar to any surgery, these procedures require sufficient preparative α-blockade.26 External-beam radiation has been particularly useful in treating osseous metastases but can provoke hypertensive crises.26 In patients with disease not amenable to surgery but with acceptable overall clinical status, 131I-MIBG can be used for locoregional control, provided 123I-MIBG imaging demonstrates good avidity.26,131,132
In patients with symptomatic unresectable disease for which other modalities have failed or are not indicated, cytotoxic chemotherapy can be considered.26,132 Cyclophosphamide-dacarabzine–based regimens are the most thoroughly evaluated.72 Three nonrandomized studies have shown radiographic and symptomatic responses in 40% to 50% of patients treated with these regimens, and 22-year follow-up of one nonrandomized prospective study published similar results with cyclophosphamide, vincristine, and dacarbazine.131 After therapy is discontinued, however, refractory disease tends to recur.26 Cytotoxic chemotherapy has also been used in borderline resectable cases, with sufficient response allowing patients to proceed with surgery.72 During treatment surveillance, imaging with CT, MRI, and bone scan as indicated every 3 months to evaluate response is recommended.131
MANAGEMENT OF LOCAL AND LOCOREGIONALLY ADVANCED ADRENOCORTICAL CARCINOMA
Surgery is the cornerstone of management for ACC in stages I to III and should always follow the principles of an oncologic resection, which requires careful preoperative planning with an appreciation for the rapid growth potential of aggressive ACCs.70,71,133 Preoperative imaging can underestimate tumor size by as much as 40%, and relationships to surrounding structures are subject to change during the interval between imaging and operation.70,133 The surgeon should be prepared for concomitant resection of liver, kidney, spleen, pancreas, stomach, colon, diaphragm, or vena cava, as suggested by imaging. Although there is no evidence that prophylactic nephrectomy of an uninvolved ipsilateral kidney improves survival, threshold for en bloc resection should be low, and nephrectomy is indicated for involvement of renal capsule or vein.71,72 The celiac axis and root of the superior mesenteric artery may be partially encased by tumor, which may be a contraindication to surgery if resection and reconstruction is not feasible.133
Locally advanced tumors of questionable resectability or behavior can be considered for systemic preoperative therapy with close monitoring. Patients with rapidly progressive disease may not derive sufficient benefit to justify a large resection, whereas tumors that respond to therapy may subsequently be deemed resectable.72,133
Adrenalectomy is traditionally performed through a midline or subcostal incision, depending on preoperative assessment of need for multivisceral resection.70 A thoracoabdominal incision may provide better exposure for diaphragmatic resection and repair or in those with prior abdominal surgery.133,134 Access to the retrohepatic inferior vena cava is necessary if it is infiltrated by tumor directly or harbors tumor thrombus requiring venotomy or resection.133 On rare occasions, median sternotomy and cardiopulmonary bypass are necessary for extracting tumor thrombus extending into the right atrium.135
The role of locoregional lymphadenectomy is not established, but one study suggests improvements in staging and possibly survival.9,72,136 Lymph node metastasis is not uncommon in ACC, and en bloc lymphadenectomy, when feasible, to clear nodes of the celiac axis, superior mesenteric artery, and renal hilum has been advocated.70,137
Consideration of laparoscopy for resection of potentially malignant low-stage ACC <8 cm is advocated by some, provided an R0 resection can be achieved and the operation is conducted by an expert surgeon.72 This issue is highly controversial, and failure to achieve an R0 resection can rarely be overcome by additional operations or adjuvant treatments.70,72,133 Independent of approach, strict adherence to oncologic principles is necessary for optimal outcome, including a complete and systematic evaluation of the entire peritoneal cavity, full mobilization of overlying uninvolved organs, and en bloc resection including the entire retroperitoneal fat pad surrounding the tumor without directly manipulating the tumor to prevent rupture of the fragile capsule and consequential peritoneal seeding.70,71,133,138 Surgical specimens should be submitted to pathology carefully marked and intact, and clips should be placed in the tumor bed for targeting of adjuvant radiotherapy if necessary.70
Patients undergoing adrenalectomy at high-volume centers have better oncologic outcome; therefore, referral to an experienced facility is advised.20,70,102,139
SURVEILLANCE AND ADJUVANT THERAPY FOR ADRENOCORTICAL CARCINOMA
Recommendations for surveillance and adjuvant therapy depend on behavior and appearance of the tumor, stage at presentation, and operative findings. Pathologic findings should be carefully reviewed as it has been reported that 25% of patients diagnosed with stage III disease by pathologic evaluation after resection were thought pre- and intraoperatively to have a lower-stage tumor.133
Data from the National Cancer Database show that 19% of patients undergoing surgery do not receive an R0 resection.137 In patients who remain good operative candidates, repeat resection by an expert surgeon should be considered, especially after an R2 resection.140 Those who are not candidates for reoperation can be considered for radiotherapy.71
In those who do undergo R0 resection, rates of local recurrence remain high at 19% to 34%, with the overwhelming majority of recurrences occurring in the first 5 years.20,70,141 Close surveillance is mandatory, and a complete evaluation every 3 months for 2 to 3 years has been advocated. Subsequently, this can be reduced to biannually until 5 years and annually thereafter. Evaluation in this setting should include a history and physical exam, a complete blood count and metabolic profile, including liver enzymes, as well as steroid profile, and cross-sectional imaging of the chest, abdomen, and pelvis.71,72 Steroid profiles should be carefully evaluated as recurrence of preoperative patterns of elevation raise concern for recurrence.21 Additional laboratories are necessary for monitoring mitotane therapy in patients receiving this treatment.142 18FDG-PET scan is not a standard modality for surveillance but is useful in characterizing newly discovered lesions on surveillance imaging as ACCs are almost invariably PET-positive.71,72,88
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


