Disorders of adrenal function occur at all ages and may be an isolated problem or one of multiple organs involved in certain syndromes. Principles that are helpful in evaluating the most common pediatric clinical situations in which an adrenal disorder is suspected include the following:
Patients with early pubic hair development (premature pubarche) will have in order of frequency:
Premature adrenarche
Precocious puberty
Late-onset congenital adrenal hyperplasia (CAH)
Adrenal or gonadal tumor
Patients with “simple” obesity are distinguished from those with Cushing syndrome by the following clinical features:
Cushing syndrome often develops rapidly.
Cushing syndrome patients do not grow well in height, but patients with obesity generally have accelerated height growth.
Cushing syndrome patients often have signs of androgen excess.
Patients with adrenocortical insufficiency will have either:
Primary adrenal failure, usually with symptoms of both glucocorticoid and mineralocorticoid (ie, salt loss) deficiency. Most commonly seen:
CAH (ambiguous genitalia in females)
Autoimmune Addison disease (often associated with other endocrinopathies)
Adrenoleukodystrophy (ALD)
Lack of adrenocorticotropic hormone (ACTH) stimulation of cortisol secretion, with symptoms of glucocorticoid deficiency alone. Most commonly seen:
ACTH deficiency associated with multiple pituitary hormone deficiencies
Suppression of the hypothalamic-pituitary-adrenal (HPA) axis due to recent discontinuation of corticosteroid treatment
The adrenal cortex secretes three classes of biologically active steroid hormones: glucocorticoids (cortisol), controlled by the HPA axis; mineralocorticoids (aldosterone), controlled through the renin-angiotensin system; and androgens (dehydroepiandrosterone [DHEA] and androstenedione), controlled by an as yet uncharacterized trophic hormone.
Metabolic effects of steroid hormones are mediated by interaction with a specific cellular receptor. The ligand-activated steroid-receptor complex dimerizes, enters the nucleus of target cells, binds to various transcription factors, and forms stable complexes with specific “steroid response elements” that then modulate transcription of specific target genes to exert the hormone effects (see Chapter 1).
The embryologically and functionally distinct adrenal medulla secretes catecholamines, mainly epinephrine (adrenaline), stimulated by the sympathetic nervous system and by high cortisol levels from the adrenal cortex. The effects of epinephrine are mediated through adrenergic receptors, which produce tissue-specific effects.
The adrenal glands lie at the upper pole of each kidney. Ectopic nodules (rests) of adrenocortical tissue derived from embryonic mesenchymal cells may be located in the broad ligaments of the female or in the testes or spermatic cords of the male. The size of the adrenal glands varies with age, mainly due to changes within the adrenal cortex (Figure 5-1). During gestation and at birth, the adrenal glands are much larger relative to body size than during adulthood, and at birth the weight of the glands is nearly that of adult glands (8-12 g). During the first year of life, the adrenal cortex undergoes involution of its fetal component reducing its size by more than 50%. The adrenal glands then gradually increase in size throughout childhood and adolescence. The structure of adrenal circulation facilitates exposure of the medulla to high glucocorticoid concentrations, which stimulate adrenal medullary epinephrine secretion (see section Disorders of the Adrenal Medulla).
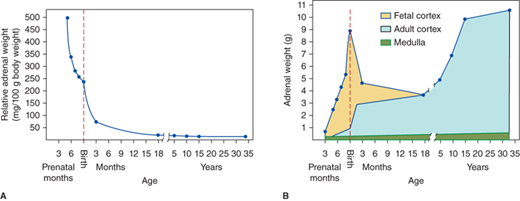
The adult adrenal cortex is divided into three histologically distinct zones: the outer zona glomerulosa, the middle zona fasciculata, and the inner zona reticularis. The zona glomerulosa is site of mineralocorticoid synthesis. There is a gradual transition from glomerulosa to the zona fasciculata, which comprises up to 75% of the cortex and functions in cholesterol storage. There is a clear transition from fasciculata to zona reticularis cells with relatively lipid-free cytoplasm. The cells of the zona reticularis and the clear cells at the interfacebetween the zona fasciculata and zona reticularis are the site of synthesis of glucocorticoids and androgens. The three zones of the adult cortex are mature by 18 to 20 years of age, and show regression associated with aging by 41 to 50 years.
The adrenal medulla is a component of the sympathoadrenal-neuroendocrine system with chromaffin cells (pheochromocytes) arranged in nests and cords, surrounded by a rich vascular network, and their abundant cytoplasm contains catecholamine storage granules. Sympathetic ganglion cells are also present. Epinephrine is the major synthesized and stored catecholamine of the adrenal medulla.
The adrenal cortex is derived from mesodermal tissue, the medulla from neuroectoderm. During the fifth week of gestation, mesothelial cells migrate into the underlying mesenchyme (near the developing gonad) and form the fetal zone of the adrenal cortex. A second migration of mesothelial cells arrive to form the smaller definitive (adult) zone. The adrenal cortex is actively synthesizing and secreting steroid hormones during the first trimester in response to tropic hormone stimulation. This is evidenced by androgen overproduction and subsequent ambiguity of the external genitalia in female infants with steroid 21-hydroxylase (CYP21A2) deficiency. During the seventh week of gestation, primitive sympathetic nerve cells differentiate into pheochromoblasts and migrate into the adrenal gland. By the 20th week, these cells have formed clusters of chromaffin cells within the adrenal medulla that does not become a distinct organ until the fetal cortex undergoes atrophy. However, it is functioning at birth as evidenced by the presence of both epinephrine and norepinephrine in the plasma and urine of newborns.
Mechanisms that lead to the differentiation of the adrenal cortex include major effects of transcription factors known as steroidogenic factor 1 (SF-1) and dosage-sensitive sex reversal adrenal hypoplasia congenital (AHC) on the X chromosome gene 1 (DAX-1). SF-1, a member of the nuclear hormone receptor family of zinc finger transcription factors, influences tissue-specific expression of steroidogenic enzymes, the ACTH receptor, the steroidogenic acute regulatory protein (StAR), and müllerian inhibiting substance (MIS). During embryogenesis, SF-1 transcripts are present in the gonads, adrenals, anterior pituitary, and hypothalamus. SF-1 regulates the expression of hormones that are critical for both adrenal functional development and male sexual differentiation, as well as other levels of the endocrine axis such as pituitary and hypothalamic function. SF-1 defects are known to cause congenital adrenal insufficiency as well as disorders of sex development (DSD).
DAX-1, an atypical member of the nuclear receptor family, is the gene responsible for X-linked AHC, in which there is impaired development of the definitive (adult) zone of the adrenal cortex and hypogonadotropic hypogonadism. Both SF-1 and DAX-1 genes are expressed in many of the same sites during embryogenesis, including the gonads, adrenal cortex, pituitary gonadotropes, and the ventromedial hypothalamus, and that DAX-1 is stimulated by SF-1.1
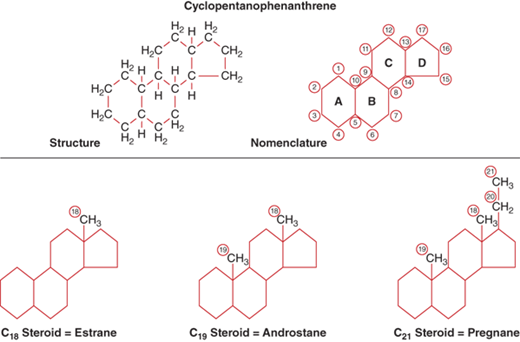
The precursor for all adrenal and gonadal steroid hormones is cholesterol. All steroids, therefore, have the same basic structural backbone, the cyclopentanophenanthrene molecule (Figure 5-2). The C18 derivative of this molecule is estrane, C19 is androstane, and C21 is pregnane. Adrenal steroids are derived from the 21- and 19-carbon rings. In general, the C19 steroids are androgens, and when there is a ketone group added at carbon 17 during metabolism, they are excreted in the urine as 17-ketosteroids. The C21 steroids are the mineralocorticoids and glucocorticoids. When a hydroxyl group is added at carbon 17, they are excreted in the urine as 17-hydroxycorticosteroids.
The stepwise conversion of cholesterol to adrenal steroid hormones involves a series of enzymes (Figure 5-3), all but one of which belong to the cytochrome P450 family of mitochondrial and microsomal proteins. The adrenal P450s require nicotinamide adenosine dinucleotide phosphate (NADPH) as an electron donor but have differing cofactor requirements. The regulation of adrenal P450 enzymatic activities depends on tropic hormone stimulation, activity of SF-1, physiological demands (eg, intravascular volume, serum sodium concentration, blood glucose), and the gradients of oxygen and steroids across the capillary bed as it passes through the three cortical zones.2
Figure 5-3
Biosynthesis of adrenocortical steroids.
Enzyme names:
CYP11A1= P450scc; activities: 20-hydroxylase, 22-hydroxylase, 20,22-lyase
CYP17A1 = P450c17; activities:a 17α-hydroxylase;b 17,20-lyase*
3β-HSD2; activities: 3β-hydroxysteroid dehydrogenase type 2; D5D4-isomerase
CYP21A2 = P450c21; activity: 21-hydroxylase
CYP11B1 = P450c11; activity: 11β-hydroxylase
CYP11B2 = P450-Aldo; activities:c two activities are 18-hydroxylase (CMO I); 18-dehydrogenase (CMO II)
CMO I = corticosterone methyl oxidase type I
CMO II = corticosterone methyl oxidase type II
POR = P450 Oxidoreductase
*CYP17 17,20 lyase activity: The conversion of 17-hydroxyprogesterone to androstenedione appears to occur only to a minor degree in normal humans. However, this reaction is enhanced in patients with CAH due to 21-hydroxylase deficiency due to the extremely high concentration of 17-hydoxyprogesterone as the substrate. In vitro, the balance of CYP21A2 and CYP17 activities within the microsomes is related to the concentration of each enzyme. Higher concentration of one enzyme reduces the activity of the other.
(Adapted, with permission, from Donohoue PA. The adrenal gland and its disorders. In: Kappy MS, Allen DB, Geffner ME, eds. Principles and Practice of Pediatric Endocrinology. Springfield, IL: Charles C. Thomas Publishers Ltd.; 2005:357-485.)
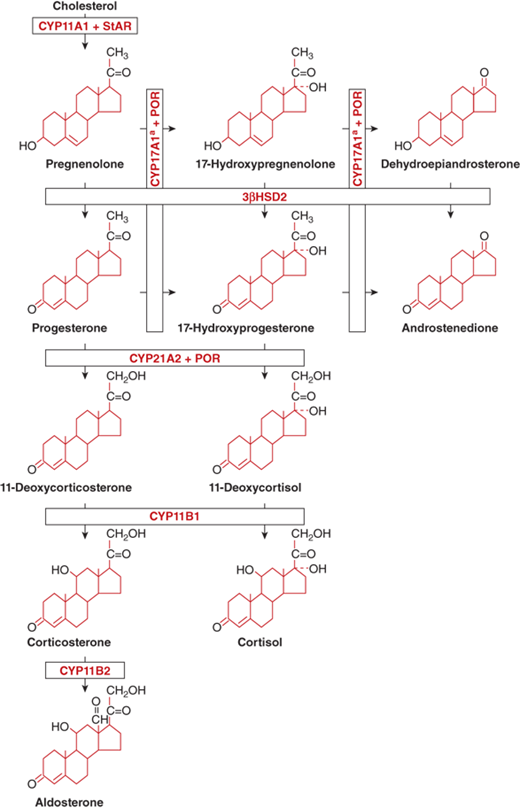
Conversion of cholesterol to pregnenolone— It is the first and rate-limiting step in adrenal steroidogenesis, mediated by the gene product of CYP11A1 (P450scc, side-chain cleavage), a single enzyme that mediates the three chemical reactions that convert cholesterol to pregnenolone: 20α-hydroxylation, 22-hydroxylation, and cleavage of the cholesterol side-chain bond at position 20-22 (see Figure 5-3). CYP11A1 is under the tropic stimulation of ACTH from the anterior pituitary gland, and requires an intermediate protein, StAR to facilitate access of cholesterol to the enzyme. StAR is expressed in the adrenal cortex and gonads, but not in the placenta (see in the section ACTH Physiology Control of Adrenal Steroid Secretion).
3a-Hydroxysteroid dehydrogenase—3β-HSD mediates the conversion of pregnenolone to progesterone and of 17-hydroxypregnenolone to 17-hydroxyprogesterone (see Figure 5-3). This enzyme system requires NAD and is located within the microsomes (endoplasmic reticulum). It is the only steroidogenic enzyme that is not a cytochrome P450, and it is required for the synthesis of glucocorticoids, mineralocorticoids, and sex steroids. There are two highly homologous human 3β-HSD isoenzymes: type 1 and type 2. Type 2 3β-HSD enzyme localizes to the adrenal cortex and gonad and is altered in 3β-HSD-deficiency CAH.
17α-Hydroxylase and 17,20-lyase reactions—They convert pregnenolone to 17-hydroxypregnenolone and progesterone to 17-hydroxyprogesterone (17OHP), both of which require hydroxylation at the 17α-position. The subsequent conversion of these two 17-hydroxylated compounds to DHEA and androstenedione, respectively, require 17,20-lyase activity (see Figure 5-3). Both 17α-hydroxylase and 17,20-lyase are enzymatic activities of the same protein, encoded by the gene CYP17A1 or P450c17. CYP17A1 is required for glucocorticoid and sex steroid synthesis, but not for mineralocorticoid synthesis. CYP17A1 is a microsomal cytochrome P450 found in both the human adrenal cortex and testis. The contribution of ACTH to physiologic CYP17A1 transcription remains unclear. CYP17A1 requires an electron transporting cofactor, P450 Oxidoreductase or POR, for normal activity.
21-Hydroxylase—It is mediated by the steroid CYP21A2 enzyme (P450c21) (see Figure 5-3), and it converts 17-hydroxyprogesteroneto 11-deoxycortisol and progesterone to 11-deoxycorticosterone (DOC). CYP21A2 is required for the synthesis of both glucocorticoids and mineralocorticoids, but not for the synthesis of androgens. CYP21A2 activity was the first biological function attributed to a cytochrome P450. CYP21A2 is a microsomal cytochrome present in all three zones of the normal adrenal cortex. ACTH stimulation increases concentrations of CYP21A2 messenger RNA (mRNA), but additional factors control CYP21A2 transcription within the functionally different zonae glomerulosa and reticularis, including SF-1. CYP21A2 also requires the electron transporting cofactor, P450 Oxidoreductase or POR, for normal activity.
11a-Hydroxylase, 18-hydroxylase, and 18-oxidase—Within the zona glomerulosa DOC undergoes hydroxylation at the 11β-position by CYP11B1 (P450c11) to form corticosterone, which is then converted by 18-hydroxylase (corticosterone methyl oxidase type I [CMO I]) to form 18-hydroxycorticosterone. This product is subsequently converted by 18-oxidase (CMO II) to form aldosterone, the major circulating mineralocorticoid. CMO I and CMO II activities reside within the enzyme CYP11B2 or aldosterone synthase. Within the zona fasciculata, 11-deoxycortisol is converted by CYP11B1 to form cortisol (see Figure 5-3). CYP11B1 and CYP11B2 are mitochondrial cytochromes. As with the above P450s (scc, c17, and c21), CYP11B1 activity and transcription are stimulated in vitro by ACTH and by SF-1.
In contrast to cells that secrete peptide or catecholamine hormones, steroid-producing cells do not maintain reservoirs of stored hormone. In addition, steroid hormones, once formed, are highly lipophilic and thus freely permeable to the cell membrane, and specific secretory processes have not been identified. As a result, de novo production of steroids from cholesterol is vital in controlling steroid hormone concentrations. The regulation of steroidogenesis can be viewed at either the level of the organism, with input of complex feedback loops involving the hypothalamus, pituitary, and kidney, or at the level of the adrenocortical cells, where production of steroids is controlled by the specific biochemical events.
Secretion of glucocorticoid by the adrenal cortex is under the control of pituitary ACTH, a single peptide chain of only 39 amino acids. Its half-life in blood is only 7 to 12 minutes. The 1,24 N-terminal amino acid sequence of ACTH has the same steroidogenic activity as the native peptide, and is the peptide sequence of synthetic ACTH used for diagnostic testing. ACTH is one of the differentially spliced products of the proopiomelanocortin (POMC) gene, as described elsewhere.
The hypothalamic factor corticotropin-releasing hormone (CRH) increases ACTH secretion. CRH is a 41 amino acid peptide, stimulated by multiple factors including a diurnal clock, low cortisol levels, and various stresses. CRH activity is located in discrete parts of the hypothalamus, particularly the median eminence. CRH is now known to be an important modulator of body size, not only through its control of ACTH and cortisol secretion but also through its direct anorexigenic effects. Several other peptides homologous to CRH, called urocortins, act in the same manner to regulate food intake.3 Although CRH is the major factor controlling ACTH secretion, other secretagogues are known, such as arginine vasopressin, insulin-induced hypoglycemia, and pyrogen-induced fever. Vasopressin and CRH have a synergistic effect on ACTH release. CRH secretion is inhibited by norepinephrine and stimulated by serotonin.
Temporally, ACTH stimulates steroidogenesisbiphasically. The acute response (seconds to minutes) reflects increased translocation of cholesterol to the inner mitochondrial membrane, where the gene product of CYP11A1 carries out the initial steps in steroidogenesis. This translocation system is induced by cyclic adenosine monophosphate (cAMP), the second messenger for most actions of ACTH, and requires ongoing protein synthesis, suggesting that a labile protein mediates the stimulation. The chronic phase (hours to days) response of steroidogenic cells to trophic hormones largely reflects increased transcription of the steroid hydroxylases in two major areas of gene regulation: cell-selective and hormonally induced expression. Cell-specificity refers to distinct but overlapping patterns of P450 enzyme expression. CYP11A1 is expressed in all classic steroidogenic tissues (adrenal cortex, testicular Leydig cells, ovarian theca cells, and placenta). The CYP17A1 gene likewise is expressed in multiple steroidogenic tissues. In contrast, CYP21A2, CYP11B1, and CYP11B2 are expressed only in the adrenal cortex, with the latter two genes displaying zone-specific expression. Second, the induction of steroid hydroxylase gene transcription is an essential component of the chronic response to trophic hormones, and several different promoter elements and transcriptional regulators have been proposed to confer responsiveness to ACTH.
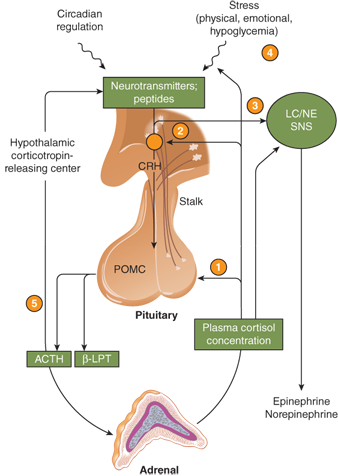
Glucocorticoid secretion is controlled by the HPA axis, as illustrated in Figure 5-4. The control of ACTH secretion is in part determined by the concentration of blood cortisol, the major circulating glucocorticoid. Cortisol in the circulation is 95% protein bound; 90% is bound to corticosteroid-binding globulin (CBG) or transcortin. Transcortin has a high affinity for cortisol as well as progesterone and can bind prednisolone and aldosterone, but not dexamethasone. The free fraction of cortisol is biologically active and is about 8% of the total cortisol when serum concentrations are normal. Compounds that bind to transcortin (eg, prednisone) will displace cortisol. At normal serum concentrations, the half-life of endogenous cortisol is 60 to 80 minutes; this may increase up to 2 hours at higher concentrations. Cortisol, both free and bound, is distributed rapidly and widely throughout the body compartments, including the extracellular fluid.
Figure 5-4
The hypothalamic-pituitary-adrenal axis. The main sites for feedback control by plasma cortisol are the pituitary gland (1) and the hypothalamic corticotropin-releasing center (2). Feedback control by plasma cortisol also occurs at the locus coeruleus/sympathetic system (3) and may involve higher nerve centers (4) as well. There may also be a short feedback loop involving inhibition of CRH by ACTH (5). Hypothalamic neurotransmitters influence CRH release; serotoninergic and cholinergic systems stimulate the secretion of CRH and ACTH; α-adrenergic agonists and γ-aminobutyric acid (GABA) probably inhibit CRH release. The opioid peptides β-endorphin and enkephalin inhibit, and vasopressin and angiotensin II augment, the secretion of CRH and ACTH. β-LPT, β-lipotropin; LC, locus coeruleus; NE, norepinephrine; POMC, proopiomelanocortin; SNS, sympathetic nervous system. (Reproduced with permission from W Arlt: Disorders of the adrenal cortex. In: Jameson JL, ed. Harrison’s Endocrinology. New York, NY: McGraw-Hill; 2013.)
The sulfation and glucuronidation metabolism of all steroid hormones occurs mainly in the liver, from which they return to the circulation through the hepatic vein to be excreted mainly by the kidney (∼90%), or excreted into the intestinal lumen through the bile. The kidney also acts as a site of the inactivation of cortisol to cortisone. Tetrahydro metabolites have been historically measured as urinary 17-hydroxycorticosteroids (17-OHCS), although the routine use of the test has been replaced by measurement of serum concentrations of specific steroids.
Glucocorticoid actions are mediated by interaction of naturally occurring and synthetic (pharmacological) steroid hormones with the glucocorticoid receptor (GR) or type 1 steroid hormone receptor. Genetic variation within the GR gene may confer increased or decreased glucocorticoid sensitivity.4
Glucocorticoid effects on carbohydrate metabolism are widespread (Table 5-1) and, at physiologicconcentrations, counterbalanced by effects of other hormones. The term “glucocorticoid” denotes effects on production and storage of glucose, so-called anti-insulin or glucose-sparing actions. Cellular glucose uptake and utilization are inhibited and glycogen synthesis stimulated. Amino acid and free fatty acid substrates for gluconeogenesis are mobilized from plasma and muscle proteins, leading to increased urinary nitrogen excretion and induction of urea cycle enzymes. In states of glucocorticoid excess, these effects may produce insulin resistance and hyperglycemia.
Biochemical Effects Increased gluconeogenesis Increased glycogen synthesis Increased blood glucose concentration aNegative nitrogen balance Lipid Metabolism Effects Increased sensitivity to lipid-mobilizing agents aNet mobilization of extremity fat and deposition oftruncal fat Gastrointestinal Effects Increased production of gastric pepsin Increased production of gastric HCl Inhibition of vitamin D–mediated calcium absorption Reticuloendothelial System Effects Granulocytosis (demargination) Decreased production of inflammatory cytokines aInvolution of lymphatic and thymic tissue Connective Tissue Effects aDecrease in collagen content of skin, bone (osteoporosis) aDecrease in collagen content of blood vessel walls (capillary fragility) aInhibition of granuloma formation Vascular Effects Increased renal plasma flow and GFR Increased free water clearance Neurologic Effects Increased cardiac conduction velocity Decreased electroconvulsive threshold Inhibition of ACTH and CRH release Inhibition of ADH release aSleep disturbances aStimulation of appetite aNeuropsychiatric alterations (psychosis, depression) aEuphoria Other Effects Requirement of normal growth aMuscle weakness aInhibition of skeletal growth aInhibition of thyroid hormone release |
Glucocorticoids exert a lipolytic action, which produces substrates for gluconeogenesis and glycogen synthesis. In hypercortisolemia, however, lipogenesis and obesity are mediated by the combined actions of excess cortisol and insulin, which stimulate appetite and produce the characteristic change in body fat distribution (central obesity, “buffalo hump”).
In addition to metabolic effects, other glucocorticoid effects include anti-inflammatory and immunosuppressive actions via direct effect on cells of the immune system, inhibition of wound healing, skeletal growth retardation and loss of bone mineral, decreased vitamin D–mediated calcium absorption from the gut, psychiatric disturbances such as depression and psychosis, and fluid and electrolyte disturbances that may be mediated through the mineralocorticoid or type 2 steroid hormone receptor.
Renin is a proteolytic enzyme that converts angiotensinogen in the liver to angiotensin I, which has mild vasopressor properties. Renin secretion from the renal juxtaglomerular cells is controlled by a baroreceptor system of the arterioles located in their proximity. Proteolytic cleavage of angiotensin I by angiotensin-converting enzyme (ACE) in the lungs produces the octapeptide angiotensin II that is a potent vasoconstrictor (Figure 5-5) and stimulates production of the major adrenal salt-retaining steroid, aldosterone. An increase in aldosterone augments increased sodium and water retention with increased intravascular volume. The resultant stretching of the renal arterioles results in decreased secretion of renin. Inversely, a decrease in blood volume as can occur in dehydration, hemorrhage, or low-salt diet will increase renin secretion.
Figure 5-5
The proteolytic cleavages of the renin-angiotensin system are illustrated. The partial amino acid sequence of angiotensinogen, and the complete sequences of angiotensin I and II are shown. The sites of formation and action of the various components of the renin-angiotensin-aldosterone system are also presented.
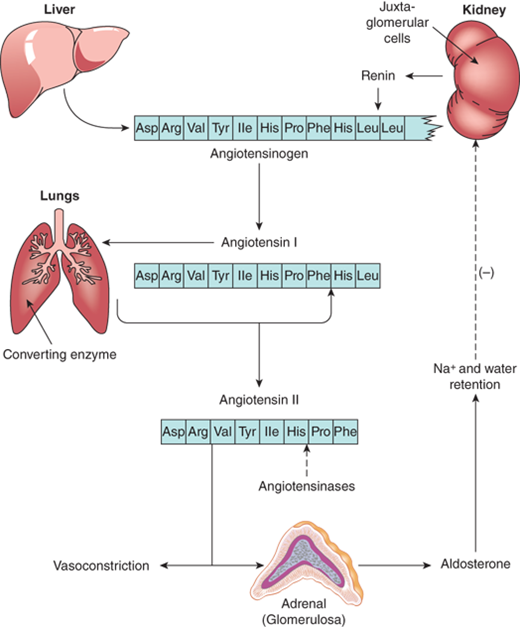
Although angiotensin II is the major stimulator of aldosterone secretion, ACTH has a definite but temporary effect as well. By contrast, prolonged and constant ACTH stimulation results in a sustained increase in plasma cortisol concentrations, but a progressive fall of aldosterone concentrations as the renin-angiotensin system is suppressed by aldosterone release and mineralocorticoid effect of the high cortisol levels.
Potassium also directly increases secretion of aldosterone by the zona glomerulosa. Although the most potent mineralocorticoid is aldosterone, some mineralocorticoid activity is found in DOC. Circulating aldosterone is bound to transcortin with 10% of the affinityof cortisol and has a plasma half-life of approximately 45 minutes. Virtually aldosterone that enters the hepatic circulation is converted to two primary metabolites, tetrahydroaldosterone and aldosterone-18-glucuronide, which are excreted in the urine.
Mineralocorticoid actions are mediated by the interaction of steroid hormones with the mineralocorticoid receptor (MR) or type 2 steroid receptor. In humans, the major natural mineralocorticoids are aldosterone and DOC. However, the MR binds also glucocorticoids such as cortisol and corticosterone with affinities similarto aldosterone. Within the major mineralocorticoid target site, the kidney, tissue-specific 11β-HSD type 2 promotes the efficient conversion of cortisol to inactive cortisone, thus removing cortisol as a significant ligand of the MR in that tissue.
Mineralocorticoids control the excretion of electrolytes, mainly in the kidney. Through ligand-receptor complex stimulation of specific ion transport channels, aldosterone enhances cortical collecting duct reabsorption of sodium, increases chloride and water retention, and thereby expands extracellular volume. Defects in these subtypes of chloride channels can cause aldosterone resistance (pseudohypoaldosteronism). Within the outer portion of the medullary collecting duct, aldosterone enhances proton excretion, particularly potassium ions. Stimulation of potassium excretion is dependent on sufficient sodium intake. Sodium conserving and potassium wasting effects of aldosterone are also present in salivary and sweat glands, and in the gut.
Effects of mineralocorticoid excess (eg, in primary hyperaldosteronism) include hypokalemia, metabolic alkalosis, and hypertension. Whereas hypokalemia and hypervolemia seem natural consequences of mineralocorticoid excess, the basis for metabolic alkalosis is unclear. It is postulated that severe potassium deficiency may alter normal acid excretion by the renal tubule.
The secretion rate of cortisol and adrenal androgens are divergent. Prepubertal children secrete cortisol at a rate (corrected for body size) similar to that of adult subjects, but adrenal androgens are almost undetectable during childhood and increase markedly at puberty without any change in ACTH or cortisol secretion. Cortisol secretion also remains similar with adult aging, while the concentrations of adrenal androgens in blood fall. In premature adrenarche, when adrenal androgens prematurely reach adult concentrations resulting in appearance of pubic hair, the cortisol secretion remains normal for size. The identity of a peptide-regulating adrenal androgen secretion remains elusive.
The adrenal androgens are DHEA, its sulfated form DHEA-sulfate (DHEA-S), and androstenedione. Both compounds exert their biological effects through extra-adrenal conversion to the potent androgens testosterone and dihydrotestosterone (DHT), which then interact with the androgen receptor (AR) within androgen target tissues (Figure 5-6A). Human steroidogenic enzymes also catalyze steps in an alternative pathway of DHT production (Figure 5-6B) referred to as a “backdoor pathway.”5 This pathway has clinical relevance in 17-hydroxylase deficiency CAH.6
Figure 5-6
General metabolism and distribution of adrenal and sex steroids. (A) Classic pathway of sex steroid synthesis and metabolism: DHEA and its sulfate DHEA-S, androstenedione, and estrone. Androstenedione is metabolized peripherally to testosterone and dihydrotestosterone, whereas estrone is metabolized to estradiol. Enzyme activities: 3β-HSD2, 3β-hydroxysteroid dehydrogenase type 2; CYP17A1, 17α-hydroxylase/17,20-lyase; ARO, aromatase; HSD17B3, 17-ketoreductase; 5α-RED2, 5α-reductase2; (Adapted with permission from Donohoue PA. The Adrenal Gland and its Disorders. In Kappy MS, Allen DB, Geffner ME (ED). Principles and Practice of Pediatric Endocrinology. Springfield, IL: Charles C. Thomas Publishers Ltd.; 2005: 357-485.) (Continued)
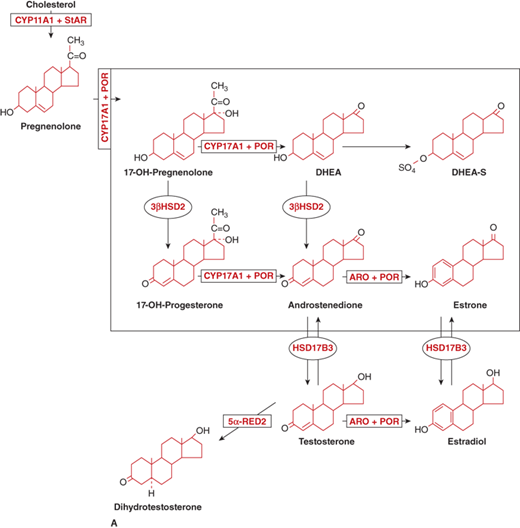
Figure 5-6
(B) Alternative pathways of androgen synthesis—additional enzymes and intermediates include 5α-RED1, 5α-reductase1; 5α-DHP, 5α-dihydroprogesterone; AKR (aldo-oxo-reductase) enzymes C2 and C4 are 3α-reductase subtypes; RoDH, 3-hydroxyepimerase. (Adapted from Flück CE et al, The American Journal of Human Genetics 89(2):201-218, 2011.6)
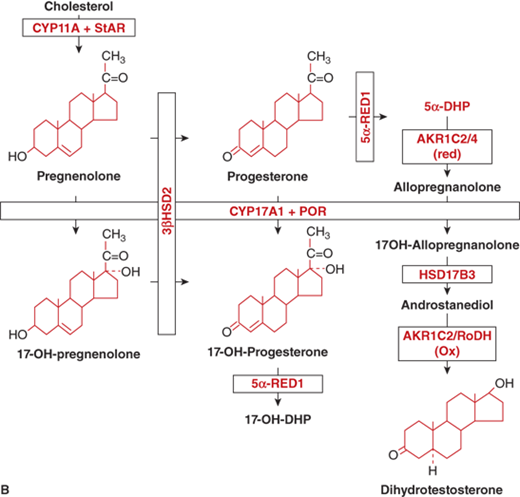
Circulating androstenedione and DHEA bind to sex-hormone–binding globulin (SHBG) to a much greater degree than to transcortin, but to a lesser degree than testosterone and DHT binding to SHBG. The plasma half-life of DHEA is relatively brief at approximately25 minutes. DHEA-S, produced in both liver and adrenal cortex, has a relatively long half-life of 8 to 11 hours and undergoes very little hepatic extraction or renal clearance. Adrenal androgens are metabolized to some degree in the liver; however, a significant degree of production and interconversion of the sex steroids occur in gonads, skin, and adipose tissue.
In adults, 15% of the circulating androstenedione is produced by peripheral conversion of DHEA and testosterone. The remainder is produced by relatively equal contributions from the adrenal and the gonad. Androstenedione is reversibly converted to testosterone by the enzyme HSD17B3 (17-ketosteroid reductase). Androstenedione may also be converted to estrone by aromatase (Figure 5-6A). Measurement of urinary 17-ketosteroids (17-KS), once commonly used as an assessment of adrenal androgen production, measures conjugated and unconjugated C19, 17-keto compounds; DHEA and androstenedione are the major precursors of the urinary 17-KS, whereas testosterone and its metabolites contribute very little.
The physiologic roles of DHEA and androstenedione in their native state remain controversial. DHEA treatment does not significantly alter perimenopausal symptoms or body composition in the elderly, but in women with adrenal insufficiency, improvement in sexualinterest and sense of well-being are reported. Effects on insulin sensitivity, body fat stores, lipid profiles, energy metabolism, and insulin-like growth factor levels remain unresolved.
The fetal adrenal is deficient in 3β-HSD, and yet cortisol production is evident by 10 weeks of gestation. This is made possible by passage of 3β-hydroxysteroids (and their sulfates) from the fetus to the placenta, which has abundant 3β-HSD and sulfatase (Figure 5-7). Progesterone and 17-hydroxyprogesterone produced in the placenta return to the fetus and are converted to cortisol and aldosterone, respectively.
Figure 5-7
Biosynthesis of steroids in fetal adrenal gland and in placenta. The fetal adrenal lacks 3β-hydroxysteroid dehydrogenase, and the placenta is rich in this enzyme. Fetal pregnenolone and 17-hydroxypregnenolone are converted to progesterone and 17α-hydroxyprogesterone in the placenta and then returned to the fetal adrenal where they serve as substrates for the formation of cortisol and aldosterone. Note that the substrate for placental estriol is fetal 16-OH DHEA. (S), sulfate; 17-OHPreg, 17-hydroxypregnenolone; 17-OHProg, 17α-hydroxyprogesterone; DHEA, dehydroepiandrosterone; 16-OHDHEA, 16-hydroxyDHEA; E1, estrone; E2, estradiol; E3, estriol.
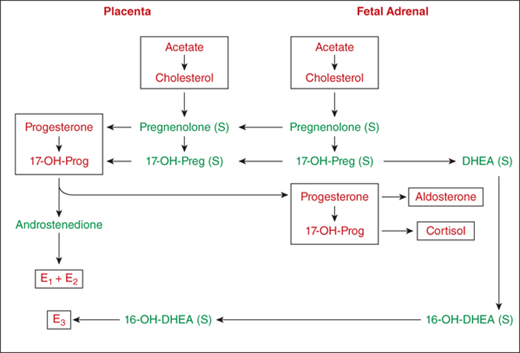
Cortisol preparations administered to the mother only minimally cross the placenta early in gestation because only unbound cortisol can be transferred to the fetus. In addition, both placenta and fetus have 11β-HSD type 2 (11β-HSD2) activity that forms inactive cortisone from cortisol. The mother contributes approximately 20% of the total fetal cortisol and 75% of fetal cortisone, the balance being secreted by the placental-fetal unit. As a consequence, to accomplish glucocorticoid therapy for the fetus (eg, CAH), it is necessary to administer a steroid-like dexamethasone that does not bind to transcortin and is not readily metabolized.
From mid-pregnancy to term, fetal plasma cortisol levels (∼2 μg/dL) are much lower than in the mother. In contrast, newborn concentrations of aldosterone are about twice those of the mother.
The relative deficiency of 3β-HSD activity in normal fetal adrenals results in the formation of large amounts of DHEA-S and its 16-hydroxy derivative. The 16-OH-DHEA is transferred into the placenta where sulfatase and aromatase transform 16-OH-DHEA into estriol. Whereas estrone and estradiol arise mainly from placental synthesis, estriol has its main origin in the fetal adrenal (see Figure 5-7). This explains why a drop in urinary estriol in pregnancy suggests either abnormal placental function, or poor fetal adrenal function.
There is little or no diurnal variation of glucocorticoid concentrations in early infancy, both in preterm and full-term infants. Studies of healthy infants show that a diurnal pattern of cortisol secretion is unlikely before 1 month of age and that most infants show a normal diurnal pattern by about 6 months of age. The appearance of the diurnal pattern seems to correlate with the development of a normal night/day sleep/wake cycle.7,8
As the fetal adrenal cortex lacks sufficient activity of some of the enzymes required for efficient cortisol secretion, it is not surprising that plasma cortisol concentrations in preterm infants, especially in response to stress and illness, are in the range considered “deficient” in full-term and older infants. The adrenal cortex of preterm infants continues to produce the major products of the fetal cortex, and without the placenta it lacks the substrates needed for sufficient cortisol secretion. Urinary excretion of “fetal zone” steroid metabolites that persists until the postconceptual age approaches 40 weeks suggests that involution of the fetal zone, and development of the definitive zone is not accelerated by premature birth. In preterm infants, there is a gestational age-related decrease in inactive cortisone concentrations relative to cortisol and a lower cortisol and 17-hydroxyprogesterone response to ACTH at less than 30 weeks’ gestation, when compared to 30 to 33 weeks. In sick preterm infants on ventilators, the cortisol response to ACTH stimulation is lower than in gestational age matched healthier infants.9
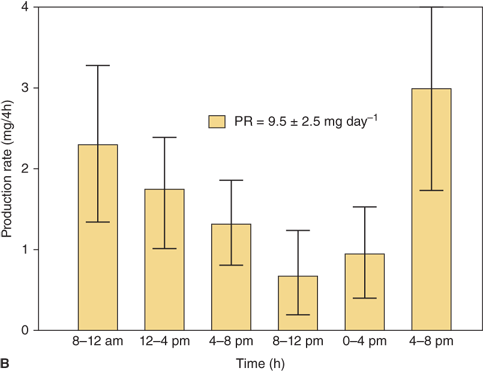
Knowledge of the cortisol production rates in normal subjects forms the basis for cortisol replacement dosage recommendations in patients with adrenal insufficiency. In normal subjects of various ages, cortisol production increases with body size. In the analysis of subjects of all ages, when the values are corrected for body surface area, the average ± SD is 12.1 ± 1.5/m2/24 h (Figure 5-8A). In the first week of life the values are somewhat greater than those of the children and adults, coming down to that range after 3 to 4 weeks of age. In children older than infants and toddlers, the cortisol production rate is estimated at 6.8 ± 1.9 mg/m2/24 h (Figure 5-8A).
Figure 5-8
(A) Cortisol production rate corrected for body surface area in normal subjects at various ages including infancy. The rates are constant throughout life except shortly after birth when they are slightly higher. (Data from Kenny FM, Preeyasombat C, Migeon CJ. Cortisol production rate. II. Normal infants, children and adults. Pediatrics. 1966;37:34.) (B) Cortisol production rate relative to body surface area in children and adolescents (From Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. J Pediatr. 1990,117:892-896, with permission.)
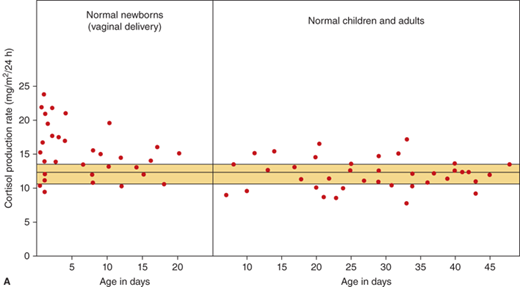
During the first 10 days of life in full-term infants, the cortisol secretion rate corrected for body size is about 1.5 to 2 times higher than it is later in infancy, childhood, and adulthood (see Figure 5-8A). In contrast, the aldosterone secretion is the same from infancy to childhood and adulthood. The concentrations of aldosterone in infancy tend to be higher than those observed later on in childhood.
Plasma adrenal androgens (DHEA, DHEA-S, androstenedione) and urinary 17-ketosteroids are relatively high shortly after birth but fall to low concentrations after 4 to 6 weeks of age, reflecting involution of the fetal zone of the adrenal cortex. In addition, plasma levels of 17-hydroxyprogesterone are high during the first 24 hours of life. It is important, therefore, to obtain a newborn screen sample after 24 hours of age to minimize the risk of a false-positive screen for CAH.
During childhood, puberty, and adulthood, cortisol secretion rate corrected for body surface area remains constant and blood cortisol concentrations follow their expected diurnal variation (Figure 5-9; see Figure 5-8). Aldosterone secretion rate and plasma concentrations are similar in childhood, puberty, and adulthood. In childhood, adrenal androgen secretion is quite low.
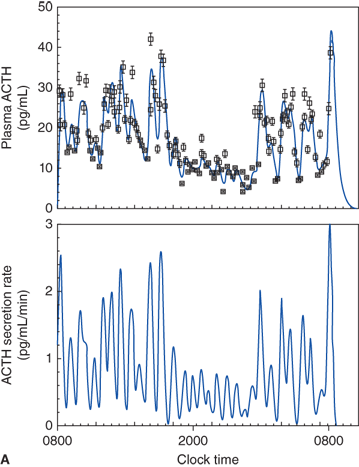
Figure 5-9
Diurnal pattern of ACTH and cortisol secretion.(Top panel: Reproduced, with permission, from Veldhuis JD, Iranmanesh A, Johnson ML, et al. Amplitude, but not frequency, modulation of adrenocorticotropin secretory bursts gives rise to the nyctohemeral rhythm of the corticotropic axis in man. J Clin Endocrinol Metab 1990;71(2):452-63. Bottom panel: Reproduced with permission from Weitzman ED, Fukushima DK, Nogeire C, et al. Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J Clin Endocrinol Met. 1971;33(1):14-22.)
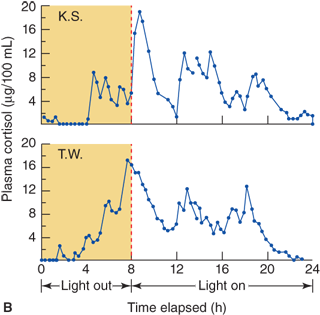
In the few years prior to gonadal maturation and puberty, under the stimulation of an as yet uncharacterized substance, there is increased secretion of DHEA, DHEA-S, and androstenedione with no change in ACTH concentration in blood or cortisol secretion rate corrected for body size (Figure 5-10) and this state is called adrenarche. As seen with the timing of gonadal puberty, adrenarche occurs 1 to 2 years earlier in girls than in boys. In both sexes the rise in plasma levels of adrenal androgens occurs before physical evidence of androgen effects are noted. In children with hypopituitarism, a lack of adrenarche is often noted. This leads to the supposition that there is a pituitary or hypothalamic factor that stimulates adrenarche. Premature adrenarche is the state in which adrenal androgen maturation occurs much earlier than gonadal maturation, producing signs of androgen effect before the expected age.
Figure 5-10
Concentrations of plasma androstenedione, dehydroepiandrosterone (DHEA), and dehydroepiandrosterone sulfate (DHEA-S) at various ages in male and female subjects. Shaded areas are means ± SD. (Redrawn from Forest MG, dePeretti E, Bertrand J. Testicular and adrenal androgens and their binding to plasma proteins in the perinatal period. J Steroid Biochem. 1980;12:25. Forest MG, Sizonenko PC, Cathiard AM, Bertrand J et al. Hypophysogonadal function in human beings during the first year of life. J Clin Invest. 1974;53:819. dePeretti E, Forest MG. Unconjugated dehydroepiandrosterone plasma levels in normal subjects from birth to adolescence in humans: the use of a sensitive immunoassay. J Clin Endocrinol Metab. 1978;47:572.)
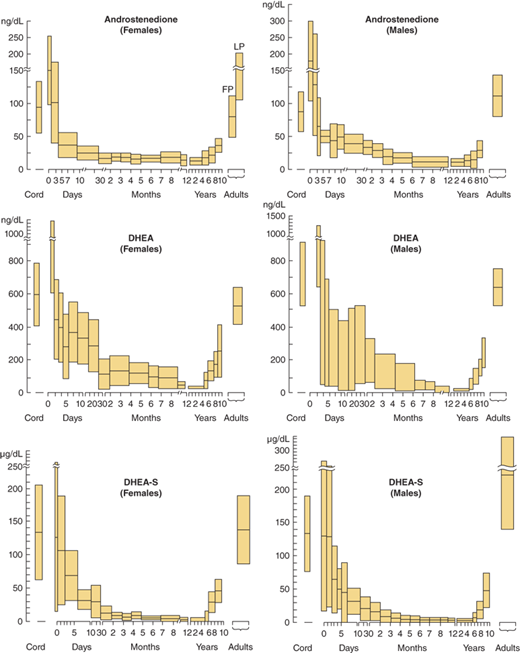
Premature adrenarche, more common in girls, is characterized clinically by appearance of pubic and/or axillary hair owing to premature adrenal androgen secretion before age 8 years in the absence of breast development. There remains controversy in the actual age that defines “premature” development. Premature sexual hair growth, body odor, and mildly increased height velocity, and/or skeletal age are typical. In the majority of children with premature adrenarche, the etiology is unknown. While most children go on to experience normal puberty, some appear to be at increased risk for early development of metabolic syndrome and (in girls) polycystic ovary syndrome (PCOS) (ovarian hyperandrogenism).10 It is unknown at this time whether the premature adrenarche itself or its association with high body mass index (BMI) or certain ethnic groups confers the increased risk of insulin resistance and ovarian hyperandrogenism.
Box 5-1 lists the circumstances in which a patient with premature adrenarche should be referred to a pediatric endocrinologist.
Box 5-1. When to Refer
Premature Pubic Hair Development
Children who are younger than the typical ages forpremature adrenarche (eg, < 6 years in girls or 8 years in boys).
Children who have advanced bone age or rapidly progressive signs of adrenarche.
Children who have abnormal elevations of sex steroid or cortisol precursor levels.
Children with a family history of congenital adrenalhyperplasia.
Cortisol—During pregnancy, estrogen-stimulated increases in transcortin concentration cause greater binding capacity for cortisol, a rise in plasma concentrations of total cortisol, and an increase in its half-life. Unbound cortisol concentrations are also elevated. At the same time, cortisol secretion is decreased during pregnancy, so that replacement therapy in pregnant women with hypoadrenocorticism typically consists of usual dosages. At the time of labor, cortisol therapy should be increased to stress levels in pregnant women with hypoadrenocorticism.
Aldosterone—Pregnant women have increased plasma renin activity (PRA), plasma aldosterone concentrations, and aldosterone secretion rate, likely reflecting a stimulation of the renin-angiotensin-aldosterone system due to a progesterone-mediated salt-losing tendency. Unlike cortisol, aldosterone does not have a specific binding protein; its binding does not increase significantly during pregnancy and its half-life in blood does not change markedly.
Androgens—Maternal DHEA-S concentrations are decreased in pregnancy. The cause for this long-described phenomenon is unknown but is postulated to be associated with increased 16-hydroxylase activity leading to the production of estriol.
Stressful conditions are often associated with increased adrenocortical secretion of cortisol, secondary to increases in CRH and ACTH output. The increase in cortisol levels with stress is a mediator of the adrenal medullary catecholamine response to stress. Stress conditions can be classified into surgical (trauma, anesthesia, tissue destruction), medical (acute illness, infection, fever, hypoglycemia), and emotional (psychological). It is not clear whether the mechanism stimulating adrenal function is unique or different for each type of stress.
Regarding infection, hormones of the HPA axis interact with components of the immune system in a complex manner. The cytokine interleukin-1 (IL-1) is secreted by macrophages in response to microbial invasion, inflammation, immunologic reaction, and tissue injury. IL-1 then activates and increases T cells, accelerates the transformation of B cells into plasma cells, which then produce antibodies. In addition, IL-1 activates the CRH-ACTH-cortisol system; the increased cortisol concentration in plasma results in a negative feedback on the macrophages. Other cytokines including tumor necrosis factor-α (TNF-α) and IL-6, as well as IL-1, stimulate CRH release but are inhibited by cortisol. The endocrine immune system interactions are influenced reciprocally by the autonomic nervous system.11
Products of the POMC gene, collectively calledmelanocortins (ACTH, α-, β-, and γ-melanocyte stimulating hormone [MSH]) interact with specific receptors on leukocytes. These receptors mediate both immunomodulatory and anti-inflammatory effects independent of production of cortisol. In addition, there are central neurogenic anti-inflammatory melanocortin-stimulated signals that utilize both adrenergic and cholinergic pathways.
Obesity related to high caloric intake is usually accompanied by an increase in cortisol secretion rate, concomitant decrease in cortisol half-life, and normal plasma concentrations. Urinary free cortisol excretion can be elevated, but when corrected for body surface area or creatinine excretion, values usually normalize. In the minority (∼20%) of obese subjects with values that remain elevated after correction for body size, other tests to rule out Cushing syndrome may be necessary.
Investigation of cortisol/cortisone metabolism in obesity has resulted in new insights. The enzyme 11β-HSD type 2 converts cortisol to inactive cortisone mostly in the kidney, whereas 11β-HSD type 1 catalyzes reactivation of cortisone to cortisol mostly in liver and adipose tissue. These two enzymes are functionally similar but are encoded by different genes on different chromosomes.
Low-calorie or protein intake is associated with elevated plasma cortisol levels. This results partly from increased secretion, and also from increased half-life due to low metabolic rate. Cortisol response to ACTH stimulation is normal, but response to CRH is blunted. Absence of cushingoid features suggests a degree of cortisol resistance. Following rehabilitation, plasma cortisol concentrations normalize.
Hypothyroidism results in slower metabolism and hyperthyroidism results in faster metabolism of cortisol, but patients with thyroid disorders tend to maintain normal plasma cortisol concentrations by either decreasing cortisol secretion in hypothyroidism or increasing it in hyperthyroidism. It should be noted that cortisol deficiency may be masked by hypothyroidism, and restoring such patients to the euthyroid state may precipitate symptoms of adrenal insufficiency. Therefore in a patient with hypopituitarism, cortisol replacement should be initiated prior to thyroxine replacement.
In patients diagnosed with growth hormone deficiency, it is important to test the HPA axis, as the risk of ACTH deficiency is present in any patient with one established pituitary hormone deficiency. Short children with growth hormone deficiency (and normal ACTH reserve) are compared with short children who do not have growth hormone deficiency; the diurnal patter of cortisol secretion are not different.
The liver extracts a large fraction of plasma steroids for metabolism, usually by reduction and by conjugation to glucuronic acid. Advanced hepatic failure results in decreased cortisol clearance and a prolonged half-life; thus plasma cortisol remains normal.
Because large fractions of cortisol and aldosterone metabolites are excreted in urine, renal failure causes lower excretion of steroid metabolites. However, the secretion of cortisol and its plasma concentration usually remains normal. As noted earlier, hyperkalemia in renal failure has a direct effect on increasing aldosterone secretion.
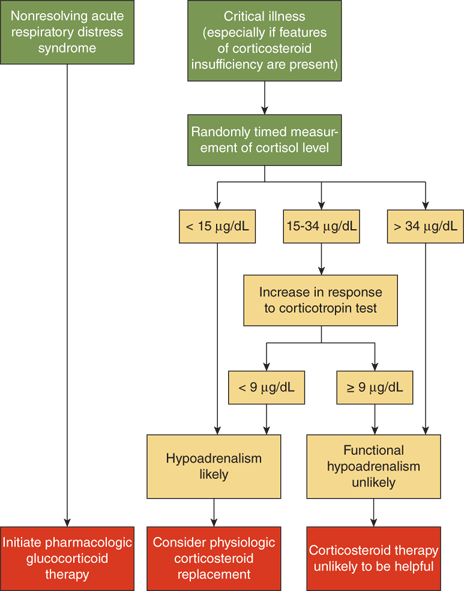
In patients with normal adrenal responsiveness to the stress of severe infection, plasma cortisol levels will increase 3- to 10-fold, unless the sepsis has caused bilateral adrenal hemorrhage. In both adults and children with septic shock, an inadequate basal cortisol level and/or an inadequate cortisol response to IV ACTH correlates highly with poor outcome. This adrenal insufficiency during stress is believed to be maladaptive and results in unchecked actions of cytokines. The lessons learned from septic shock have been expanded to include other forms of critical illness. Definite guidelines for diagnosis and treatment of cortisol deficiency in these settings have been established (Figure 5-11).12 The guidelines for the use of hydrocortisone for hemodynamic support in pediatric septic shock suggest cutoffs similar to those shown in Figure 5-11. The doses of hydrocortisone recommended for hemodynamic support in septic shock are far higher than those used for “stress coverage” in healthy patients with a history of adrenal insufficiency.13
Figure 5-11
Diagnostic algorithm for evaluation of adrenal function in the critical care setting. These guidelines outline the steps in evaluation of patients with suspected adrenal insufficiency, and have been applied to both adults and children. The cutoff values may vary slightly depending on the reference laboratory. This schema is based on using a high-dose (250 μg) rapid IV ACTH stimulation test. (Reproduced with permission from Cooper and Stewart.12)
Dichlorodiphenyltrichloroethane (DDT), its -dichloroethanederivative (DDD), and related compounds such as o,p’-DDD can suppress adrenal cortical function. The latter substance is used in the treatment of adrenal carcinoma with some success but is often poorly tolerated because of nausea, anorexia, and protracted vomiting.
Another substance, 1,1 bis-(p-aminophenyl)-1-methyl-propanone, called amphenone B, can also suppress adrenal function. This compound and o,p’-DDD tend to produce a destruction of the cells of the adrenal cortex rather than to specifically inhibit its function.
Aminoglutethimide appears to block CYP11A1, thereby inhibiting transformation of cholesterol to pregnenolone. Aminoglutethimide also has inhibitory effects on CYP11B1, reducing cortisol and aldosterone production.
The antifungal agent ketoconazole is known to generally suppress cytochrome P450 enzyme activity. This is associated with a blunted cortisol response to ACTH and thus has a risk of inducing adrenal insufficiency. It has been used therapeutically to suppress adrenal androgen excess in patients with CAH, cortisol excess in Cushing syndrome, and gonadal androgen secretion in boys with precocious puberty. Other drugs that inhibit mitochondrial P450s and thus impact steroid synthesis include fluconazole and etomidate, and potentially others that are currently under investigation.
In addition to compounds that directly affect enzymes involved in adrenal steroid biosynthesis, others have their effect at the target cell level. The aldosterone antagonism effects of spironolactone, progesterone, and 17-hydroxyprogesterone relate to their ability to compete for the MR.
Drugs associated with increased cortisol metabolism include phenobarbital, phenytoin, rifampin, and troglitazone. Peripheral resistance to glucocorticoids has been described with mifepristone through interaction with the GR, and psychotropic drugs (chlorpromazine, imipramine) can inhibit glucocorticoid-induced gene transcription. Anticoagulants may cause adrenal insufficiency indirectly by causing adrenal hemorrhage.
The progestational agent megestrol acetate (Megace), a potent appetite stimulator used in patients with cachexia, may suppress adrenal function by binding the GR and activating the negative feedback loop that inhibits ACTH and cortisol secretion. In a few patients on high doses, its interaction with the GR has caused Cushing syndrome.
Disorders of deficient or excess adrenocortical function can be diagnosed with proper selection of tests of basal HPA function and dynamic tests of HPA axis capacity.
The plasma concentrations of total and free cortisol, the main human glucocorticoid, are determined accurately using HPLC tandem mass spectrometry assays.
Pulsatile secretion and diurnal variation of cortisol: Because cortisol is secreted in a pulsatile manner, small variations can be observed when blood samples are collected at frequent intervals. The episodic secretion of ACTH and cortisol with time results in the diurnal or circadian variation (rhythm) of plasma ACTH and cortisol concentrations, with a surge of secretion between4 and 6 am, followed by decreasing concentrations to their lowest levels in the evening and during the night. Thus it is important to obtain plasma samples for analysis of basal cortisol or ACTH secretion at appropriate times given the clinical situation: usually 8 am for tests of cortisol sufficiency and/or 4 pm for tests of cortisol excess. Stressful situations are likely to raise the ACTH and cortisol concentrations, including anxiety and pain associated with phlebotomy.
The circadian rhythm is related to sleep-wake cycles. Modification of the sleep-wake distribution throughout a 24-hour period changes the profile of cortisol concentrations, as long as the modification is maintained for a sufficient time. The rhythm of cortisol can be altered by strict sleep reversal, but night workers with a variable schedule tend to maintain a normal diurnal rhythm. Blind subjects may also have disturbances of the normal diurnal pattern.
Unbound or free plasma cortisol is excreted by the kidney. As only free plasma cortisol is biologically available, the fraction of unbound plasma cortisol that is excreted in urine as free cortisol reflects glucocorticoid activity at the cellular level. Current assays show only 1% to 2% cross-reactivity with some steroids (cortisone, corticosterone, 11-deoxycortisol, and 17-hydroxyprogesterone), but prednisolone has about 30% cross-reactivity. The range of urinary free cortisol for normal adults varies by laboratory, and this range is 10 to 35 μg/24 h in females and 10 to85 μg/24 h in males. When corrected for urinary creatinine concentration, the range is approximately 8 to 40 μg/g of creatinine. In children, the range is approximately 3 to 9 μg/24 h, and when the values are correlated with body size, the range corrected for body surface area being 25 to 65 μg/m2/24 h. When corrected for urinary creatinine, the range is approximately 7 to 25 μg/g creatinine. During pregnancy the reference ranges are higher. In Cushing syndrome the concentrations are usually above 150 μg/24 h and in adrenal insufficiency less than 10 μg/24 h.
Salivary glands and sweat glands excrete only unconjugated, unbound (free) glucocorticoids. Salivary samples can be collected at home in a nonstressful setting. The diurnal pattern of salivary cortisol concentration reflects those of total plasma cortisol, but concentrations are about 90% lower. This is due in part to the metabolism of cortisol to cortisone by the salivary glands, as well as the fact that it measures free, not total, cortisol. Nighttime salivary cortisol concentrations have been validated as a useful screening test for Cushing syndrome in both adults and pediatric patients,14 and this remains its most significant clinical use. It is replacing the 24-hour urinary free cortisol as the initial screening test. A midnight salivary cortisol level greater than 0.44 mg/dL identifies pediatric patients with Cushing syndrome with 99% specificity and 100% sensitivity.15
The use of salivary cortisol levels in clinical medicine has expanded, especially in the area of neurobehavioral studies that examine responses to stress. In the setting of endocrinology, salivary cortisol is not a reliable method for diagnosing adrenal insufficiency. It has been studied as a tool to monitor replacement therapy in patients with adrenal insufficiency and CAH; however, the levels are highly variable and have not added significant information in the monitoring of these patients.16,17
Time-based reference ranges for salivary cortisol concentrations are similar in prepubertal children and adults. The approximate ranges at 0800 are 0.17 to 1.2 mg/dL; at 1600, 0.10 to 0.3 mg/dL; and at 2300, 0.03 to 0.17 mg/dL. After a 1-mg dose of dexamethasone at 2300, the following morning 0800 salivary cortisol should be less than 0.1 mg/dL.
Plasma ACTH can be measured accurately by immunometric assays. As with cortisol, it is important to standardize the time at which blood samples are collected, usually at 8 am (normal range 20-120 pg/mL) and 4 pm (0-50 pg/mL). Because cortisol has a negative feedback on ACTH secretion (ie, when cortisol concentrations are elevated, those of ACTH tend to decrease, and reciprocally), it is often helpful to measure simultaneously the cortisol and ACTH concentrations.
Assay techniques for the measurement of CRH concentration are available, but determining its concentration in plasma is not often useful. Because CRH reaches the anterior pituitary corticotrophs via a portal system of vessels, studies of CRH in peripheral circulation have not been fruitful. It is probable that there is a diurnal variation of CRH in the pituitary portal vessels, but a diurnal pattern has not been observed in plasma.
After prolonged loss of ACTH secretion, such as in states of cortisol excess or in hypopituitarism, the adrenal cortex can become resistant to its stimulatory effect.18 The rapid IV ACTH test is often used to screen for ACTH deficiency in such patients. In this setting, either a low-dose (0.1-1 μg) or high-dose (250 μg) test has been used. The 250-μg test is widely used, and the cortisol response 30 to 60 minutes after injection should peak at or above 18 to 20 μg/dL to rule out adrenal insufficiency. The 1-μg test, though, may be more sensitive in detecting milder degrees of adrenal gland suppression. The expected cortisol response to the low-dose test is reported by some authors to be slightly less than with the high-dose test19 but by others to be the same as the high-dose test.18 However, the cortisol response to insulin-induced hypoglycemia remains the gold standard, and the peak cortisol levels seen with the 250-μg ACTH test are comparable to those seen with insulin-induced hypoglycemia.19
In critically ill patients, especially those with sepsis or hemodynamic instability, screening for relative adrenal insufficiency involves measurement of blood cortisol before and 30 to 60 minutes after 250 μg of IV ACTH. In this stressful setting, basal levels of cortisol should normally exceed 30 μg/dL; if less than 15 μg/dL and if the cortisol concentration does not rise by at least 9 μg/dL over baseline values that are less than 30, relative adrenal insufficiency is suspected.
Because glucagon stimulates both ACTH and growth hormone secretion, the glucagon test can be used to assess both cortisol release and pituitary reserve of ACTH secretion. A baseline sample is drawn, followed by a 0.03-mg/kg (to a maximum of 1 mg) dose of glucagon given by SQ (or IM) injection. Serum cortisol can be measured every 30 minutes, up to 3 hours following the administration of glucagon. Patients, in particular young children, can experience abdominal discomfort, nausea, and/or vomiting during the course of the glucagon stimulation test.20 The cortisol response is similar to that seen with the ACTH stimulation tests.
Another IV administration of ovine CRH (100 μg/dose) is normally followed by an increase in plasma ACTH from approximately 8 to 25 pg/dL with a concomitant increase in plasma cortisol. The ACTH response to CRH may be useful in the differentiation of Cushing from pseudo-Cushing syndrome. Normative data now exist for ACTH response to CRH test in very-low-birth-weight infants, in whom antenatal maternal corticosteroid treatment may induce adrenal suppression.
Hypoglycemia is a potent stimulus for ACTH secretion. At 8 am after an overnight fast, 0.05- to 0.075-U/kg regular insulin is given by IV injection. Serum glucose and cortisol are measured at baseline and every 15 minutes for 1 hour. A normal response is seen if the 8 am baseline cortisol concentration is within the expected reference range, and the level doubles or rises over 20 μg/dL with hypoglycemia. Basal cortisol and the insulin tolerance test (ITT) are thought to be more accurate for detection of HPA axis deficiency than is the CRH test or the low-dose (1 μg) ACTH test; however, it is contraindicated in some patients such as infants.18
Administration of a potent glucocorticoid, such as dexamethasone, provides negative feedback suppressing ACTH secretion and secondarily decreasing cortisol secretion. There are several types of dexamethasone suppression tests (Table 5-2). One method uses a single nighttime dose of 1 mg followed by measurement of 8 am cortisol. More prolonged “low-dose” and “high-dose” tests last several days and are followed by measurement of plasma ACTH and cortisol. The test is useful in the evaluation of patients with suspected Cushing disease. Dexamethasone suppression test may also differentiate whether hyperandrogenemia in a female results from adrenal or gonadal secretion, the latter not being sensitive to ACTH suppression.
Overnight Test: 20 μg/kg dexamethasone PO (up to 1.0 mga) at 2300, with serum cortisol level at 0800 the following morning Normal response: 0800 cortisol < 5 μg/dL rules againstCushing Syndromeb,c Low-Dose Test: 48 h, 2 mg/d (0.5 mg PO q6h) Normal response: 0800 cortisol level obtained 2 h after the last dose of dexamethasone ≤ 1.8 μg/dL rules againstCushing syndromed 48 h, 2 mg/d test combined with CRH stimulation test at 0800, 2 h after the last dose of dexamethasone Normal response: serum cortisol < 1.4 μg/dL, 15 min after CRH 1 μg/kg IV (or 100 μg IV in adults) rules againstCushing syndrome High-Dose Test for Differential Diagnosis ofACTH-Dependent Cushing Syndromee: 48 h, 8 mg/d (2 mg dexamethasone PO q6h) Suppression of urinary 17-OHCS by > 50% consistent with pituitary source of ACTH Overnight test (8 mg dexamethasone PO at 2300) Serum cortisol at 0800 the mornings before and after shows that suppression of serum cortisol by > 50% suggests pituitary source of ACTH |
Nonspecific tests, such as electrolytes in serum and in urine, as well as occasionally in sweat and saliva, can aid in diagnosing abnormalities in mineralocorticoid secretion or action. However, precise immunoassays for aldosterone and PRA provide a direct assessment of mineralocorticoid secretion and its control by the renin-angiotensin system.
Plasma concentrations of aldosterone follow a circadian variation that is similar to but less marked than that of cortisol. Aldosterone concentrations change slowly (24-48 hours) in response to high- or low-sodium intake. In contrast, changes in body position (standing vs lying down) bring about rapid but transient variation in aldosterone concentrations.
In early infancy, plasma aldosterone concentrations are variable but in general are elevated relative to older children and adults. After 1 year of age and up into adulthood, mean values remain constant, although there are large individual variations.
Aldosterone secretion rate is relatively constant throughout life, so that recommended replacement doses of 9α-fluorocortisol in patients with mineralocorticoid deficiency are similar throughout childhood and adulthood. There are slighter lower values observed during the first 2 weeks after birth, but the blood levels of aldosterone are higher. Changes in sodium diet influence the aldosterone secretion rate as well as plasma concentrations of this steroid.
PRA is measured by the rate of release of angiotensin II from the renin substrate angiotensinogen. The angiotensin II being formed is measured by immunoassay and the PRA is expressed as angiotensin II formed in pg/mL of plasma per hour. PRA shows episodic variation as well as mild diurnal variation. On an average sodium intake the PRA is 10 to 50 pg/mL/h. Concentrations increase markedly during a low-sodium diet and are suppressed by a high-sodium diet. PRA may be elevated in patients who are receiving certain medications, such as ACE inhibitors, vasodilators, diuretics, and spironolactone.
Direct determination of renin concentration in plasma is made with a specific immunoassay. Normal ranges vary based on sodium intake, posture, and age.
Clinical signs suggesting hypersecretion of androgens should prompt measurement of their concentration in blood. In the prepubertal male, they include increase in size of the penis; scrotal pigmentation and thinning, pubic, axillary, and facial hair; and deepening of the voice. In the female, they include premature pubarche, enlargement of the clitoris, deepening of the voice, disruption of menstrual cycling, and hirsutism.
The main adrenal androgens include androstenedione, DHEA, and DHEA-S (see Figure 5-6A). Their plasma concentrations are typically measured by HPLC tandem mass spectrometry. These steroids do not bind to the androgen receptor and therefore have negligible biological activity. However, 5% to 10% of DHEA is converted to androstenedione, and a similar fraction of androstenedione is converted to testosterone.
Adrenal androgens have very little significance in adult males, but they contribute about 75% of the testosterone in adult women. The life cycle of plasma concentrations of adrenal androgens is shown in Figure 5-10. The concentration of DHEA-S in plasma is the best indicator of adrenarche during pubertal maturation. Among hormonal steroids in human plasma DHEA-S has by far the highest concentrations, owing to its long half-life.
Androgens, whether secreted by the adrenals or by the gonads, are partially metabolized into 19-carbon steroids with a 17-ketone and are excreted as urinary 17-ketosteroids. About one-third of testosterone produced in the body is excreted in the urine as 17-ketosteroids. The conversion of androstenedione to testosterone is a reversible reaction catalyzed by HSD17B3; thus a portion of circulating testosterone is converted back to a 17-ketosteroid. Normal values are low before puberty (< 2 mg/24 h), compared with adolescence and adulthood. Normal values in adults are higher in males than females (7-17 mg/24 h vs 4-13 mg/24 h). Although most providers prefer to measure specific plasma androgens, the determination of urinary 17-ketosteroids can still be useful in specific situations, such as when phlebotomy is not feasible or assessment of 24-hour integrated adrenal androgen production is warranted.
Disorders of hypoadrenocorticism can be classified as either intrinsic to the adrenal cortex (primary hypoadrenocorticism), due to CRH/ACTH deficiency (central or secondary hypoadrenocorticism), or due to unresponsiveness to trophic or adrenocortical hormones (Table 5-3).
Primary Adrenocortical Insufficiency Congenital adrenal aplasia X-linked adrenal hypoplasia congenita (AHC) due to DAX-1 defect Adrenal hypoplasia due to SF-1 defect Congenital adrenal hyperplasia due to CYP21A2 deficiency Congenital adrenal hyperplasia due to CYP11B1 deficiency Congenital adrenal hyperplasia due to CYP17A1 deficiency Congenital adrenal hyperplasia due to 3βHSD2 deficiency Congenital adrenal hyperplasia due to CYP11A1 or StAR deficiency Congenital adrenal hyperplasia due to P450 oxidoreductase (POR) deficiency Adrenoleukodystrophy/adrenomyeloneuropathy Wolman disease (acid lipase deficiency) Steroid sulfatase deficiency (X-linked ichthyosis) Smith-Lemli Opitz syndrome Mineralocorticoid deficiency due to CMOI or CMOII (CYP11B2) defect Chronic hypoadrenocorticism (Addison disease) Secondary to Deficient ACTH Secretion Panhypopituitarism or multiple anterior pituitary hormone deficiencies Isolated ACTH deficiency (eg, due to TPIT or POMC defect) Cessation of pharmacologic glucocorticoid treatment Resection of unilateral cortisol-secreting tumor Infants born to steroid-treated mothers Secondary to End-Organ Unresponsiveness Pseudohypoaldosteronism (mineralocorticoid resistance) ACTH resistance Cortisol resistance |
Signs and symptoms of adrenocortical insufficiency depend on the hormones that are deficient (Table 5-4), and/or in excess (eg, androgens) in patients with biosynthetic defects of cortisol and aldosterone. Clinical features of chronic hypoadrenocorticism may be influenced by other symptoms resulting from the autoimmune polyglandular syndromes (APS) (Table 5-5).
| Glucocorticoid Deficiency | Mineralocorticoid Deficiency | Adrenal Androgen Deficiencya |
|---|---|---|
| Fasting hypoglycemia | Weight loss | Decreased pubic and axillary hair |
| Increased insulin sensitivity | Fatigue | Decreased libido |
| Decreased gastric acidity | Nausea, vomiting, anorexia | |
| Nausea, vomiting | Salt-craving | |
| Fatigue | Hypotension | |
| Muscle weakness | Hyperkalemia, hyponatremia, metabolic acidosis with normal anion gap | |
| Increased Melanocyte-Stimulating Hormone | ||
| Hyperpigmentation | ||
| Type 1 | Type 2a | |
|---|---|---|
| General | ||
| Prevalence | Rare | Not as rare as type 1 |
| Age at onset | Infancy/young childhood | Adolescence/adulthood |
| Inheritance | Autosomal recessive | Dominant |
| Gender predominance | None | Female |
| Cause | AIRE gene mutations | Polygenic/multifactorial |
| HLA association | None | HLA DR/DQ association |
| Hormonal Abnormalities | ||
| Hypoparathyroidism | > 80% | Rare |
| Addison disease | > 60% | > 80%b |
| Hypogonadism | > 30% | < 10% |
| Autoimmune thyroid disease | < 10% | > 70% |
| Type 1 diabetes | < 20% | 40%-50% |
| Hypopituitarism | Uncommon | Rare |
| Other Clinical Features | ||
| Mucocutaneous candidiasis | 80%-100% | Not seen |
| Ectodermal dysplasia | > 75% | Not seen |
| Alopecia | 25%-30% | < 10% |
| Pernicious anemia | 10%-15% | Variable |
| Autoimmune hepatitis | 10%-15% | Rare |
| Intestinal malabsorption | 10%-25% | Rare |
| Vitiligo | 10% | < 10% |
| Asplenism | Uncommon | Not seen |
| Keratoconjunctivitis | Uncommon | Rare |
Treatment of adrenal insufficiency consists of hormone replacement therapy. In primary adrenal insufficiency, both glucocorticoid and mineralocorticoid treatment is usually needed. This group of patients is at risk for a salt-wasting crisis at the time of diagnosis or duringtimes of stress. In isolated cortisol deficiency (eg, due to ACTH deficiency or resistance), only glucocorticoid treatment is needed. In all patients, the dose of glucocorticoid must be increased during times of stress. A detailed description of treatment protocols is given as follows.
Causes of primary hypoadrenocorticism can be classified based on physiology and embryology (see Table 5-3). Lack of differentiation of adrenal cortex results in congenital adrenal aplasia or hypoplasia (eg, X-linked adrenal hypoplasia). Abnormalities of membrane receptors for ACTH or angiotensin II may result in inadequate cortisol (eg, congenital unresponsiveness to ACTH) or aldosterone secretion, respectively. Steroidogenic defects may affect formation of cortisol (eg, CAH) or aldosterone (eg, CMO I and CMO II deficiencies due to aldosterone synthase defects). Conditions altering the availability of cholesterol for steroidogenesis may also occur (eg, ALD, adrenomyeloneuropathy [AMN]), Wolman disease, and steroid sulfatase deficiency orX-linked ichthyosis). Finally, normal adrenal glands can be damaged or destroyed by extrinsic factors (eg, bilateral adrenal hemorrhage of the newborn, adrenal hemorrhage of acute infection, and Addison disease.)
This is a rare congenital malformation owing to a developmental disorder of the adrenal anlage. It manifests very early in life by vomiting and diarrhea with hyponatremia, hyperkalemia, and hypoglycemia. Shortly thereafter, affected children display tachycardia, hyperpyrexia, apnea, cyanosis, and eventually seizure, vascular collapse, and coma, often misdiagnosed as septicemia, intracranial hemorrhage, or other serious illness. For this reason it is often diagnosed at autopsy. Pathology often reveals small amounts of adrenal tissue either in its usual location or associated with the gonads, or scattered throughout the peritoneum. If adrenal tissue is found, the condition may be attributed to one of the other syndromes described later on.
In X-linked congenital adrenal hypoplasia (also known as adrenal hypoplasia congenita [AHC]), both cortisol and aldosterone secretions are impaired. It is described as cytomegalic adrenal hypoplasia because of the histological appearance of the adrenal cortex in patients studied at autopsy. X-linked AHC most often presents in the newborn period (1-4 weeks) with signs and symptoms of a salt-wasting crisis. However, affected patients with variable degrees of adrenocortical development may not have symptoms of adrenocortical insufficiency until later in childhood.
X-linked AHC generally occurs with hypogonadotropic hypogonadism. Mutations of DAX-1 (dosage-sensitive sex reversal adrenal hypoplasia on the X chromosome gene 1) cause combined AHC and HH in affected males. In some patients, there is a contiguous gene syndrome due to a deletion, including DAX-1 (discussed later on in the chapter), dystrophin (causing Duchenne muscular dystrophy [DMD]), and the glycerol kinase gene (causing juvenile glycerol kinase deficiency [GKD]). Patients with this complex (DMD, AHC, and GKD) also have a characteristic phenotype that may include short stature, mental retardation, testicular abnormalities (cryptorchidism, hypogonadism), and peculiar facies (drooping mouth, wide-set eyes).
Congenital defects in adrenocortical development may also be due to mutations of the SF-1 gene. This syndrome may include male to female sex reversal. Females with primary adrenal failure and SF-1 mutations have normal genitalia. Mutations of SF-1 may also lead to DSD without adrenal insufficiency.21
Treatment of congenital adrenal hypoplasia or aplasia consists of replacement therapy for both mineralocorticoid and glucocorticoid deficiencies (as described in detail elsewhere in the chapter).
This disorder is caused by a genetic abnormality of the ACTH receptor (also known as the melanocortin 2 receptor [MC2R]), leading to inability of the zona fasciculata and reticularis to produce cortisol in response to ACTH stimulation. By contrast the zona glomerulosa secretes aldosterone normally. This condition is distinct from adrenal unresponsiveness to ACTH due to developmental, autoimmune, or infectious causes.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


