Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .

Introduction
Endogenous Cushing’s syndrome (CS) is a rare disorder caused by chronic overproduction of cortisol. The most common causes of endogenous CS are adrenocorticotropin (ACTH)-producing pituitary adenoma (Cushing’s disease), ectopic ACTH-producing tumors (ectopic CS) and cortisol-producing adrenal adenoma (commonest cause of adrenal CS) [ ]. Less common causes of adrenal CS are cortisol-producing adrenocortical carcinoma and bilateral micro- and macronodular adrenal hyperplasia. Far more common than endogenous CS is exogenous CS, that is, patients who develop cushingoid features due to prolonged glucocorticoid treatment in supraphysiological doses.
CS, irrespective of the etiology, may have serious consequences on health. The risk for cerebro- and cardiovascular diseases, psychiatric illnesses, skeletal fractures, and severe infections are greatly increased in patients with CS [ ]. Consequently, mortality is increased, not only in patients with untreated hypercortisolism, but also in patients who have achieved biochemical remission following treatment. Thus, CS is a serious disorder that needs to be diagnosed and treated properly without delay.
This chapter summarizes the epidemiology, genetics, clinical characteristics, management, follow-up, and outcome in patients with adrenal CS. A special emphasis is put on the pathophysiology and treatment of hypertension, a complication seen in most of the patients, as well as cardiovascular morbidity.
Causes and epidemiology of adrenal Cushing syndrome
Approximately 20%–30% of patients with endogenous CS have adrenal CS [ , ]. Since the cortisol production in all forms of adrenal CS is autonomous, and not dependent on stimuli from the pituitary gland, these together are also referred to as ACTH-independent CS ( Table 14.1 ). The most common cause of adrenal CS is a cortisol-producing adrenocortical
| Etiologies of endogenous CS | Proportion of patients with CS |
|---|---|
| ACTH-dependent CS | |
| ACTH-producing pituitary adenoma | 60%–70% |
| ACTH-producing pituitary carcinoma | Very rare |
| Ectopic ACTH-producing tumors | 5%–10% |
| ACTH-independent (Adrenal CS) | |
| Cortisol producing adrenal adenoma | 10%–20% |
| Cortisol producing adrenal carcinoma | 5% |
| Bilateral macronodular adrenal hyperplasia | <2% |
| Bilateral micronodular adrenal hyperplasia | <2% |
The etiologies of endogenous Cushing’s syndrome are shown in Table 14.1 .
Epidemiological studies that have estimated the incidence of adrenal CS are few. In two nationwide studies, one from New Zealand [ ], and one from Denmark [ ], the annual incidence of cortisol-producing adrenal adenoma was 0.3 and 0.6 cases per million per year, respectively. Similarly, in a recent study from Sweden, the incidence of adrenal CS was 0.5 cases per million per year [ ]. In that study, 22 out of 82 (27%) patients with endogenous CS had adrenal CS; 14 had cortisol-producing adrenocortical adenoma, 5 had cortisol-producing adrenocortical carcinoma, two had bilateral micronodular adrenal hyperplasia, and one had bilateral macronodular adrenal hyperplasia. Finally, in a nationwide study from Korea, the incidence of adrenal CS was 1.3 per million per and year between 2002 and 2017 [ ]. The vast majority (94%) of 1199 patients included in the study had benign adrenal CS.
The majority (80%–90%) of patients with cortisol-producing adrenal adenoma are women [ , ]. In patients with bilateral adrenal hyperplasia, the gender distribution is more or less equal [ , ]. Cortisol-producing adrenal adenoma and bilateral macronodular adrenal hyperplasia are most often diagnosed at an age between 40 and 60 years [ , ], while the median age at diagnosis of bilateral micronodular adrenal hyperplasia is 20 years [ ].
In children, although rare, ACTH-producing pituitary adenomas are the most common cause of endogenous CS (as in adults) [ ]. However, in neonates and infants, adrenal CS is more common [ , ], mainly due to rare genetic syndromes associated with adrenal CS as discussed below.
Genetic syndromes associated with adrenal Cushing syndrome
Several genetic syndromes are associated with adrenal CS [ ].
Multiple endocrine neoplasia type 1 (MEN-1) is an autosomal dominant disorder, typically considered to be a syndrome characterized by development of hyperparathyroidism, pituitary adenoma, and pancreatic neuroendocrine tumors. However, patients with MEN-1 are also at increased risk for developing several other tumors, including adrenal tumors that affect at least 10% of individuals with this syndrome [ ]. In fact, 5% of patients with MEN-1 develop CS, that may either be ACTH-dependent due to ACTH-producing pituitary adenoma or ectopic ACTH-production, or ACTH-independent due to benign or malignant adrenocortical tumors as well as bilateral adrenal hyperplasia [ , ].
McCune-Albright syndrome is a rare genetic disorder characterized by fibrous dysplasia of the bone, café-au-lait skin pigmentation, and various endocrinopathies, including precocious puberty, hyperthyroidism, and overproduction of growth hormone [ ]. Adrenal CS is a rare but serious component of McCune-Albright syndrome, seen in 7% of the patients [ ]. CS in these patients presents in the neonatal period and may be fatal if not adequately treated [ ].
Carney complex is an autosomal dominant disorder that can present with a wide variety of features, including lentiginosis, cutaneous-, cardiac-, and breast-myxomas, thyroid and testicular tumors, psammomatous melanotic schwannomas, and acromegaly [ ]. Adrenal CS is one of the most common manifestations of Carney complex, affecting approximately 70% of the women and 20% of the men [ ]. CS in patients with Carney complex is caused by bilateral micronodular adrenal hyperplasia, also called primary pigmented nodular adrenocortical disease (PPNAD).
Other syndromes that rarely cause adrenal CS are Beckwith-Wiedemann syndrome and Li-Fraumeni syndrome [ ].
Genetics of ACTH -independent Cushing syndrome
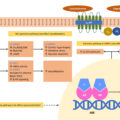
Significant progress has been made in the understanding of the genetic basis of adrenal CS [ ]. Specifically, genetic alterations resulting in abnormal function of the cAMP-dependent protein kinase A (PKA) system, a major cell-signaling pathway in humans and other species, is of great importance. This was first discovered in patients with McCune-Albright syndrome who have an activating mutation in GNAS, a gene that encodes the alpha subunit of the G-protein complex, an important regulator of the cAMP-dependent PKA system [ ]. Similarly, the majority (70%) of patients with Carney complex have a mutation in PRKAR1A , encoding another regulatory protein of PKA (Protein Kinase CAMP-Dependent Type I Regulatory Subunit Alpha) [ ]. Furthermore, mutations in PRKACA , a gene encoding the Cα subunit of PKA, have been found in 35%–65% of patients with cortisol-producing adrenal adenoma and overt CS [ ]. Thus, genetic variants responsible for activation of the cAMP-dependent PKA system, resulting in increased cell proliferation and cortisol-production, appear to be an important player in the pathogenesis of adrenal CS.
Another recent and important discovery was that one-fourth of the patients with primary bilateral macronodular adrenal hyperplasia (PBMAH) have a damaging mutation of the gene encoding armadillo repeat-containing protein 5 (ARMC5), a protein that inhibits the cell cycle and stimulates cell apoptosis in the adrenal gland [ , ]. Interestingly, patients with cortisol-producing adrenal adenoma secondary to PRKACA mutation, as well as patients with PBMAH secondary to ARMC5 mutation, both have a more severe phenotype with higher cortisol concentrations than patients with same disorders but without these mutations [ , ].
Clinical features
Patients with CS may present with various symptoms and signs. The degree of cortisol secretion varies greatly in patients with CS, from a marginally elevated concentration causing mild symptoms, to greatly increased plasma levels causing a severe phenotype. The most common symptoms are fatigue and weight gain [ , , ], where a central fat accumulation with an increased waist circumference, supraclavicular and dorsocervical fat pads, and round face are characteristic ( Table 14.2 ). Due to the presence of muscle atrophy, the extremities are often disproportionally thin compared to the rest of the body, and muscle weakness is common. Cutaneous symptoms are also common, including skin atrophy, striae, easy bruising, plethora, and acne. Most patients with CS have hypertension, which can be therapy-resistant, and one-third have diabetes mellitus. Other common symptoms are depression, anxiety, cognitive impairment, insomnia, menstrual disorders, and osteoporosis with an increased fracture risk. Some symptoms such as bluish-purple striae, facial plethora, proximal muscle weakness, easy bruising, and unexpected osteoporotic fractures are more specific for CS [ ]. These are, however, often absent in patients with mild CS.
| Clinical characteristics of CS | Proportion |
|---|---|
| Fatigue | 90% |
| Weight gain with central fat accumulation, increased waist circumference, supraclavicular and dorsocervical fat pads and round face | 80%–90% |
| Hypertension | 75%–85% |
| Skin changes including skin atrophy, blue-violet striae, easy bruising, facial plethora, hirsutism, and acne | 70%–80% |
| Proximal muscle atrophy and muscle weakness | 60%–70% |
| Cognitive dysfunction including memory and concentration defects and impaired attention | 60%–80% |
| Oligomenorrhoea or amenorrhoea | 50%–60% |
| Sleeping difficulties | 50% |
| Decreased libido and erectile dysfunction | 50% |
| Depression | 40%–60% |
| Diabetes mellitus | 30% |
| Osteoporotic fractures | 20% |
None of the symptoms of endogenous CS can be used to determine the etiology of the hypercortisolism, i.e., whether it is caused by adrenal, pituitary, or ectopic source. However, patients with ectopic CS often have a shorter disease course, and more severe hypercortisolism, characterized by severe muscle weakness, hyperglycemia, therapy-resistant hypertension, hypokalemia, and sometimes disorientation and/or psychosis. Also, due to pronounced catabolism, weight gain and the otherwise characteristic central fat accumulation may not be present in these patients with severe hypercortisolism.
Many of the symptoms and clinical signs of CS are nonspecific and are common in the general population. A long diagnostic delay is, therefore, unfortunately common, especially in patients with mild hypercortisolism. In a study from Germany, the mean time from the first contact with a physician due to symptoms caused by CS until a correct diagnosis was made was 3 years [ ]. On an average, the patients met with five different specialists before being diagnosed; most common specialists were general practitioners, gynecologists, dermatologists, internists, and/or orthopedists [ ]. In a meta-analysis, including data from more than 5000 patients with CS, the mean diagnostic delay was 38 months in patients with ACTH-producing pituitary adenoma, 30 months in patients with adrenal CS, and 14 months in patients with ectopic CS [ ]. This delay is unfortunate. The quality of life is greatly reduced in patients with CS and does not improve until successful treatment has been provided [ ]. Also, severe comorbidities such as cardiovascular disease, fractures, thromboembolic disease, and psychiatric illness are common before diagnosis [ ]. Furthermore, a long diagnostic delay is associated with increased mortality, emphasizing the importance of early diagnosis and treatment [ ].
Pathophysiology of hypertension in adrenal Cushing syndrome
Hypertension is one of the most commonly occurring features of CS, being present in 80%–85% of the patients [ , ], and seem to be equally common in patients with adrenal CS as in patients with pituitary CS [ , ]. When evaluated with 24-hour ambulatory blood pressure monitoring, most patients with CS lack the nocturnal blood pressure dip that is normally seen in healthy individuals and in patients with essential hypertension [ ]. The blood pressure in patients with untreated CS can be greatly increased. In fact, a recent study from Austria showed that 9% of patients with CS had required in-patient management for hypertensive crisis on some occasions before they were diagnosed with the syndrome [ ].
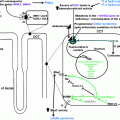
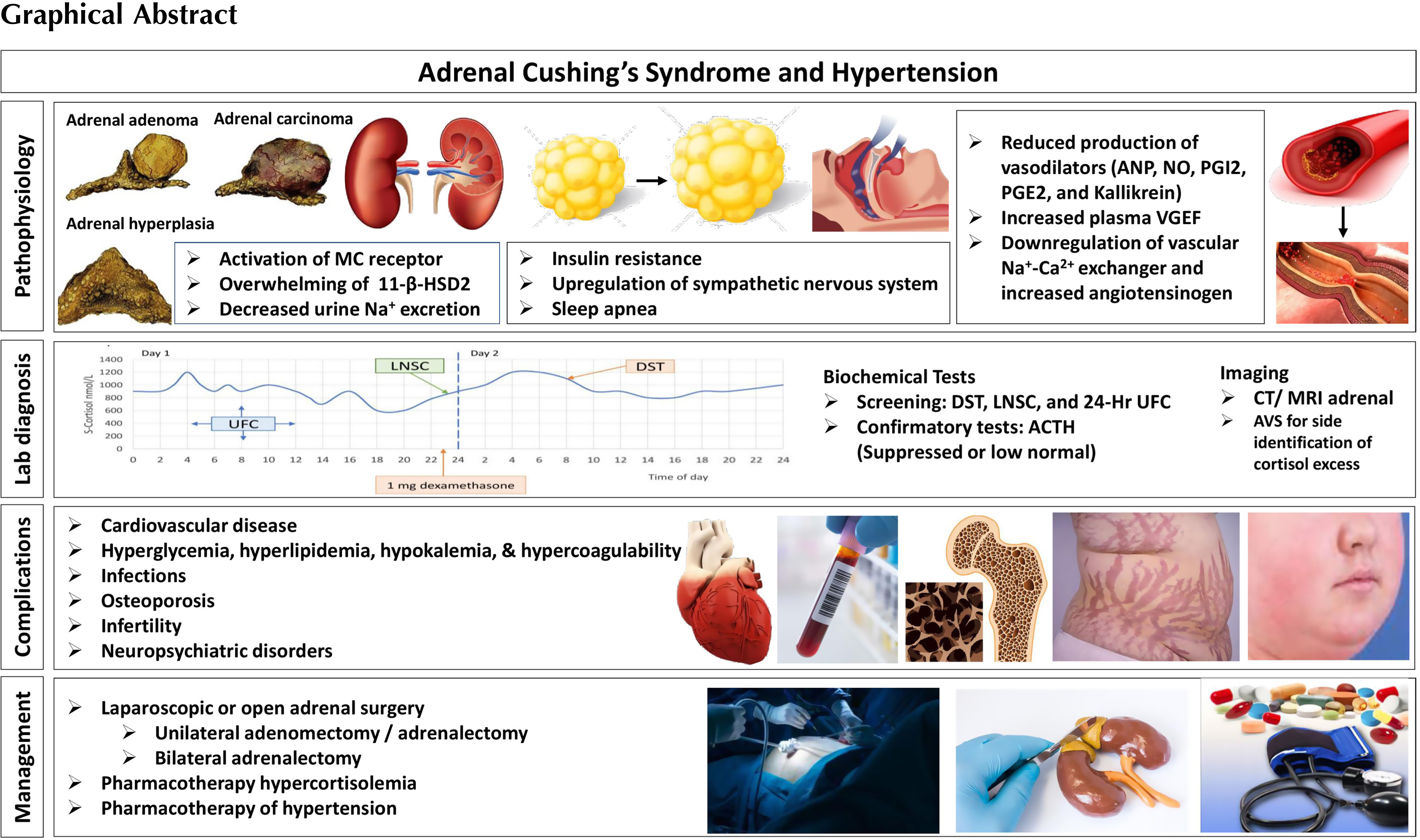
The pathogenesis of hypertension in patients with CS is not fully understood [ ]. However, one of the major pathophysiological mechanisms for the development of hypertension in patients with CS is the direct stimulatory effects of cortisol on the mineralocorticoid receptor in the kidneys ( Fig. 14.1 ). In healthy individuals the mineralocorticoid receptor is protected from the actions of cortisol by the 11β-hydroxysteroid-dehydrogenase type 2 (11β-HSD2), an enzyme that converts cortisol to its inactive form, cortisone. In patients with CS, especially in patients with very high cortisol production, the enzyme becomes saturated, allowing the excessive amounts of cortisol to activate the mineralocorticoid receptor. This, in turn, leads to sodium retention, expansion of plasma volume, and hypertension [ , ].
The activation of the mineralocorticoid receptor is not the only explanation for the development of hypertension in patients with CS, and several other factors have been suggested ( Fig. 14.1 ). However, most of the available literature on this topic dates back to the 1980s and 1990s, the studies often include a small number of subjects, and the findings are frequently based on administration of glucocorticoids to healthy subjects, rodents or in vitro cells, and not on patients with endogenous CS. Nevertheless, among suggested mechanisms are increased concentrations of angiotensinogen [ ], increased expression of the angiotensin II type 1 gene in smooth muscle cells [ ], increased vascular sensitivity to angiotensin II and/or catecholamines [ ], and increased cardiac sensitivity to catecholamines [ ]. Decreased production of vasodilating mediators have also been suggested including kallikrein, prostacyclin (prostaglandin I 2 ), prostaglandin E 2 [ , ], and nitric oxide [ , ]. Similarly, increased production of vasoconstricting mediator like enothelin-1 [ ] is also suggested. Furthermore, erythropoietin-mediated reduction of vasodilating mediators and increased vasoconstriction has also been proposed [ ].
Investigations for adrenal CS
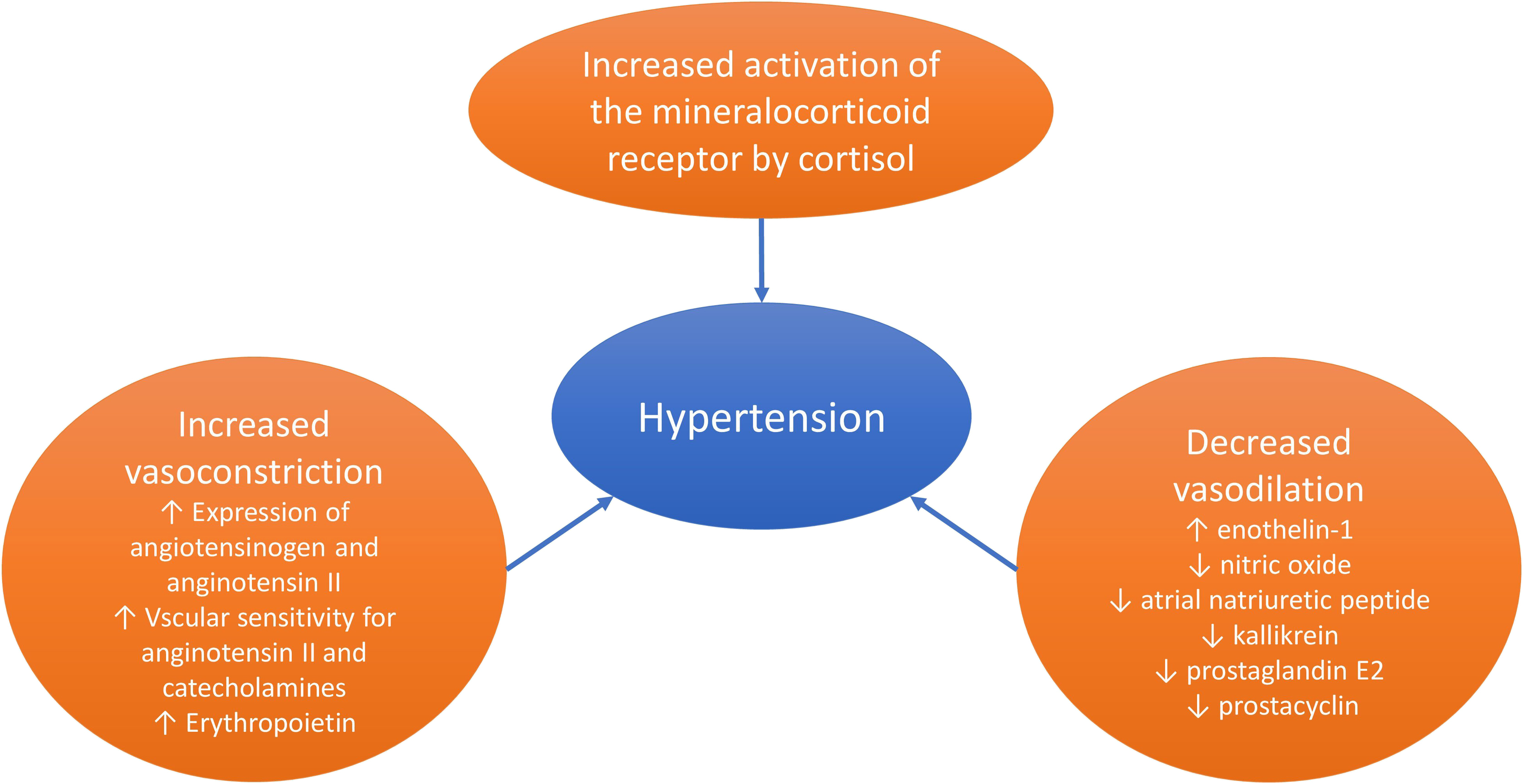
Upon clinical suspicion of CS, a screening for the disorder should be initiated without a delay. Three tests are useful for this purpose: 24-h urinary free cortisol (UFC), overnight 1 mg dexamethasone suppression test (DST), and late-night salivary cortisol (LNSC) [ , ]. With these tests, three cardinal features of endogenous CS are utilized to diagnose the syndrome, that is, the increased cortisol production (UFC), the nonsuppressible autonomous cortisol production by the administration of exogenous glucocorticoids (DST), and the lack of normal circadian cortisol rhythm (LNSC) ( Fig. 14.2 ).
Which test is chosen depends on the local availability of these tests with a validated analytical method and well-defined diagnostic cut-offs, as well as certain patient characteristics such as renal function, severity of the hypercortisolism, and the use of medications that either increase the concentrations of corticosteroid-binding globulin (e.g., oral estrogen) or induce a more rapid degradation of dexamethasone (e.g., carbamazepine) ( Table 14.3 ). All the tests have a high sensitivity for identifying patients with CS. DST has slightly higher sensitivity than the other two tests while the specificity is lower [ ]. More details on the initial diagnostic evaluation of CS are discussed in Chapter 13 : ACTH-dependent Cushing’s syndrome (Fernandes et al. 2022).
| Screening tests | Principles | Strengths | Limitations |
|---|---|---|---|
| Urinary free cortisol (UFC) | Reflects total cortisol production for 24 h |
|
|
| Dexamethasone suppression test (DST) | Reflects the autonomous cortisol production in CS that cannot be suppressed by administration of exogenous glucocorticoids | High sensitivity |
|
| Late-night salivary cortisol (LNSC) | Reflects the lack of normal circadian cortisol rhythm in CS patients with high concentrations in the evening (e.g., 10–11 p.m.) |
|
|
The next step in the investigation of patients with biochemically confirmed CS is to measure plasma ACTH level. Patients with adrenal CS have low ACTH levels, while those with ACTH-dependent CS have normal or high plasma ACTH. For patients with adrenal CS, computed tomography (CT) of the adrenal glands is primarily recommended, although magnetic resonance imaging (MRI) can also be used [ ]. The most common cause of adrenal CS, an adrenal adenoma, is almost always larger than 1 cm in diameter, and therefore easy to detect. The most important differential diagnoses in patients with adrenal CS and unilateral adrenal lesion are cortisol-producing adrenal adenoma and adrenocortical carcinoma. Some of the radiological characteristics are useful to differentiate between these two disorders. Adrenal adenomas, compared to adrenocortical carcinoma, are smaller (mean 3.5 cm, [range 2–7 cm], vs. mean 14.5, [range 7.5–21] cm), have lower unenhanced attenuation [mean 11, range −16 to +41 Hounsfield units (HU), versus mean 28, range +20 to +31 HU] and have homogenous texture [ ]. Differential diagnoses in patients with CS and bilateral adrenal lesions are bilateral adenomas, bilateral micro- and macronodular hyperplasia, and ACTH-dependent CS [ ]. Of note is that patients with bilateral micronodular hyperplasia may have morphologically normal adrenal glands on imaging.
Adrenal venous sampling in patients with ACTH-independent hypercortisolism, and either bilateral adrenal lesions or normally appearing adrenals on imaging, has been found to be useful for differentiating between unilateral and bilateral overproduction of cortisol, both in patients with overt adrenal CS and autonomous cortisol secretion (see below) [ ]. Although the studies are based on a limited number of patients, adrenal venous sampling should be considered in patients who are considered to be candidates for surgical treatment.
Treatment of adrenal CS
First-line treatment for patients with CS due to cortisol-producing adrenal adenoma is unilateral adrenalectomy, performed by either retroperitoneal or transabdominal laparoscopic technique [ ]. Adrenalectomy in these cases is always curative when performed by an experienced surgeon. Unilateral adrenalectomy is also the primary treatment for patients with cortisol-producing adrenocortical cancer. However, many patients with adrenocortical cancer have metastatic disease at the time of diagnosis, which makes curative treatment only rarely possible.
Bilateral adrenalectomy is the treatment of choice in patients with overt CS due to micro- and macronodular adrenal hyperplasia [ , ]. Recently, however, unilateral adrenalectomy where the larger gland is removed has been suggested for patients with PBMAH in a French study [ ]. In this study, all patients were considered to be in biochemical remission following surgery, and CS recurred in only two out of 15 patients during the 5 years of follow-up. In contrast, two other studies have shown a significantly higher recurrence rate; and not more than one-third of the patients were still biochemically controlled after few years of follow-up [ , ]. Similarly, recurrence is common in patients with PPNAD treated with unilateral adrenalectomy [ , ].
Cortisol-lowering medical treatment is rarely used as a long-term treatment option for patients with adrenal CS, e.g., patients with mild hypercortisolism and the elderly with multiple comorbidities [ ]. However, they are mainly used preoperatively to reduce the risk of perioperative complications (see below).
Preoperative treatment of adrenal CS
Hypertension and diabetes mellitus should be adequately controlled preoperatively in patients with CS in order to reduce the risk of complications during and after surgery. Pharmacotherapy with adrenal steroidogenesis inhibitors is effective in lowering the cortisol concentrations, and subsequently reducing the blood pressure and the plasma glucose concentrations. Preoperative cortisol-lowering treatment is also important for patients with severe hypercortisolism, especially when they present with serious comorbidities such as pronounced muscle weakness and neuropsychiatric symptoms including psychosis [ , ].
Cortisol-lowering medical therapy alone is not always sufficient in reducing the high blood pressure in patients with CS. In these cases, treatment with either of the mineralocorticoid receptor antagonists, spironolactone or eplerenone, is often useful, especially in patients with moderate to severe hypercortisolemia and/or hypokalemia. Based on studies on the pathophysiology of hypertension in patients with CS, angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers are also considered to be a good choices [ , , ], either as monotherapy or in combination with calcium channel blockers [ ]. Detailed discussion of the management of hypertension in patients with CS can be found in chapter 13 : ACTH-dependent Cushing’s syndrome.
Patients with CS have a greatly increased risk of developing deep vein thrombosis and pulmonary embolism [ , ]. In fact, in a meta-analysis of 48 studies including data from more than 7000 patients with CS, a 17-fold higher risk of perioperative venous thromboembolic events was demonstrated [ ]. Thus, administration of perioperative thromboprophylaxis with low molecular weight heparin should be considered in all patients with CS, especially in patients with moderate to severe hypercortisolism [ ].
Another serious complication in patients with CS is severe infections, caused by both commonly acquired bacteria, and by opportunistic microorganisms [ ]. In a recent study from Sweden, a 14-fold increased risk for sepsis was observed from diagnosis until one year after treatment [ ]. Also, in a large study from the European Registry on CS (ERCUSYN), the most common causes of perioperative death were infections [ ]. Prophylactic broad-spectrum antibiotics as well as prophylactic treatment against Pneumocystis jirovecii should be considered, especially in patients with severe hypercortisolism [ ].
Postoperative treatment of adrenal CS
All patients with adrenal CS who are successfully treated with unilateral adrenalectomy develop adrenal insufficiency and, therefore, need glucocorticoid replacement therapy postoperatively for several months [ ]. In a study from Germany, the median time to recovery of normal hypothalamic–pituitary–adrenal (HPA) axis function in patients treated for unilateral adrenal CS was 2.5 years [ ]. Other studies have shown that adrenal CS patients regain their normal cortisol production on an average within 6–18 months postoperatively [ , ]. In all these studies, however, 10%–20% fail to restore normal HPA axis function [ ]. Interestingly, preoperative cortisol-lowering medical treatment seems to reduce duration of postoperative adrenal insufficiency [ ]. Since adrenal insufficiency can be a life-threatening condition, it is of great importance to thoroughly counsel the patients about this possibility [ , ]. Specifically, the patients should be informed about which actions are needed in situations associated with increased physical or psychological stress, that is, the need of increased dose of oral glucocorticoids during intercurrent illness with fever as well as before minor procedures, such as tooth extractions, and the need of parenterally administrated glucocorticoids in the case of severe illness, major surgery, or adrenal crisis [ ].
Long-term outcome of adrenal CS
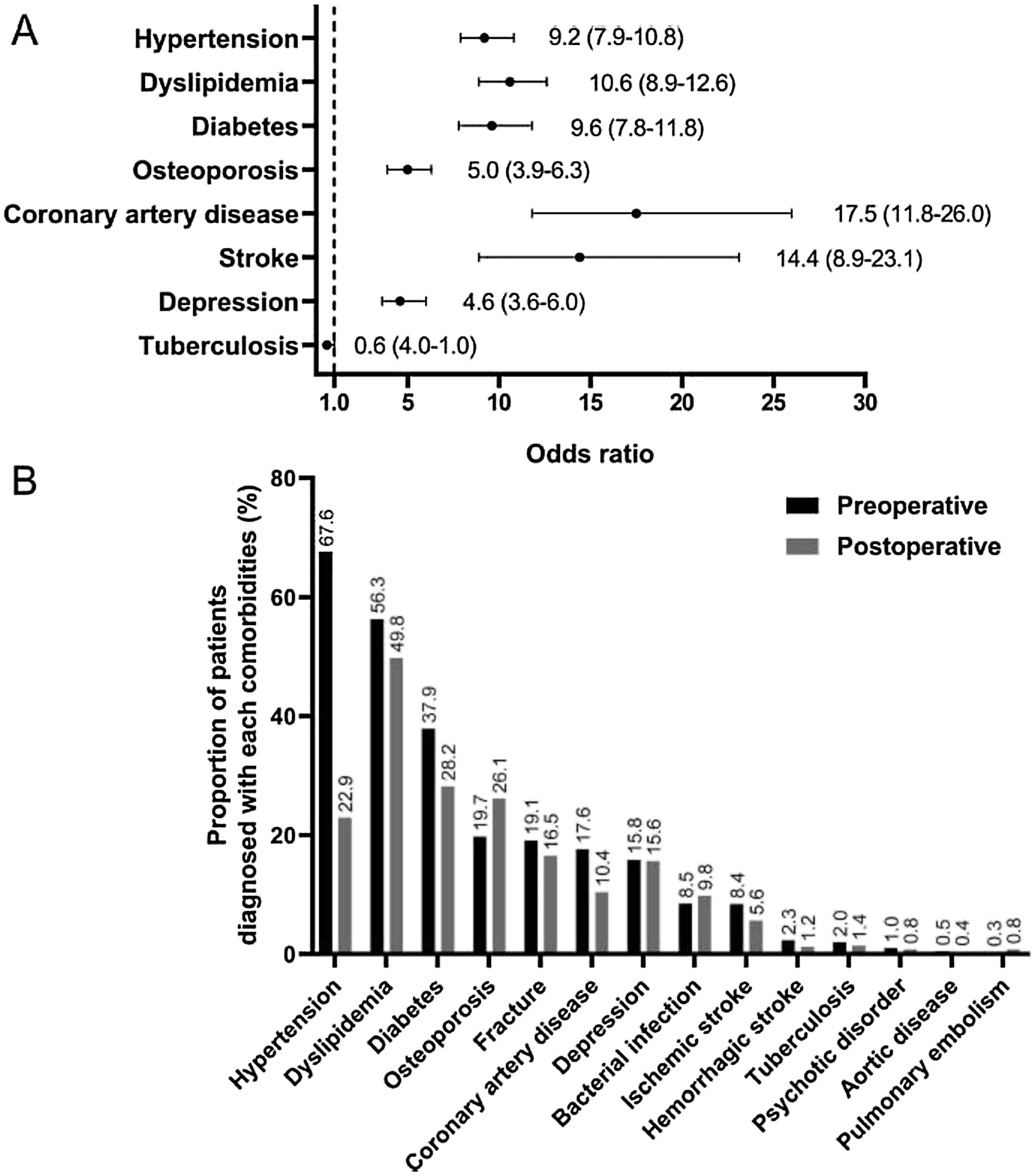
The cardiometabolic risk profile in patients with CS significantly improves following the treatment and resolution of the hypercortisolism as shown in ( Fig. 14.3 ). In a study from Italy including 14 patients with ACTH-producing pituitary adenoma, and 15 patients with cortisol producing adrenal adenoma, the prevalence of hypertension, diabetes mellitus, impaired glucose tolerance, and dyslipidemia before treatment was 72%, 24%, 27%, and 59%, respectively, compared to 10%, 0%, 10%, and 21% in controls matched for age, sex, and BMI [ ]. One year following adrenalectomy, the prevalence of hypertension in patients with adrenal CS due to cortisol producing adrenal adenoma fell from 80% to 40%, the prevalence of impaired glucose tolerance/diabetes mellitus from 53% to 7%, and dyslipidemia from 53% to 27%. In another study, including 18 patients with adrenal CS, 83% had hypertension at diagnosis, but only 17% following surgery [ ]. Interestingly, the outcome, in both studies, was better in patients with adrenal CS compared to patients with ACTH-producing pituitary adenoma [ , ]. In a recent study from the Mayo clinic, including patients with CS of various etiologies and hyperglycemia, a significant improvement in glycemic control following treatment was observed [ ].