Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .



Introduction
The human adrenal cortex has three distinct layers; the outer zona glomerulosa producing mineralocorticoids, the central zona fasciculata producing glucocorticoids, and inner zona reticularis producing androgens and some glucocorticoids [ ]. These steroid hormones alter the vascular hemodynamics and play key roles in the regulation of blood pressure and cardiovascular homeostasis. Excessive production of these hormones may lead to hypertension [ ], whereas deficiency may cause life-threatening illness including profound hemodynamic failure [ , ]. This chapter describes the physiological aspects, the biological effects, and pathophysiological alterations of adrenal cortical hormones in relation to human blood pressure regulation.
Physiological aspects
An overview of adrenal steroid synthetic pathways
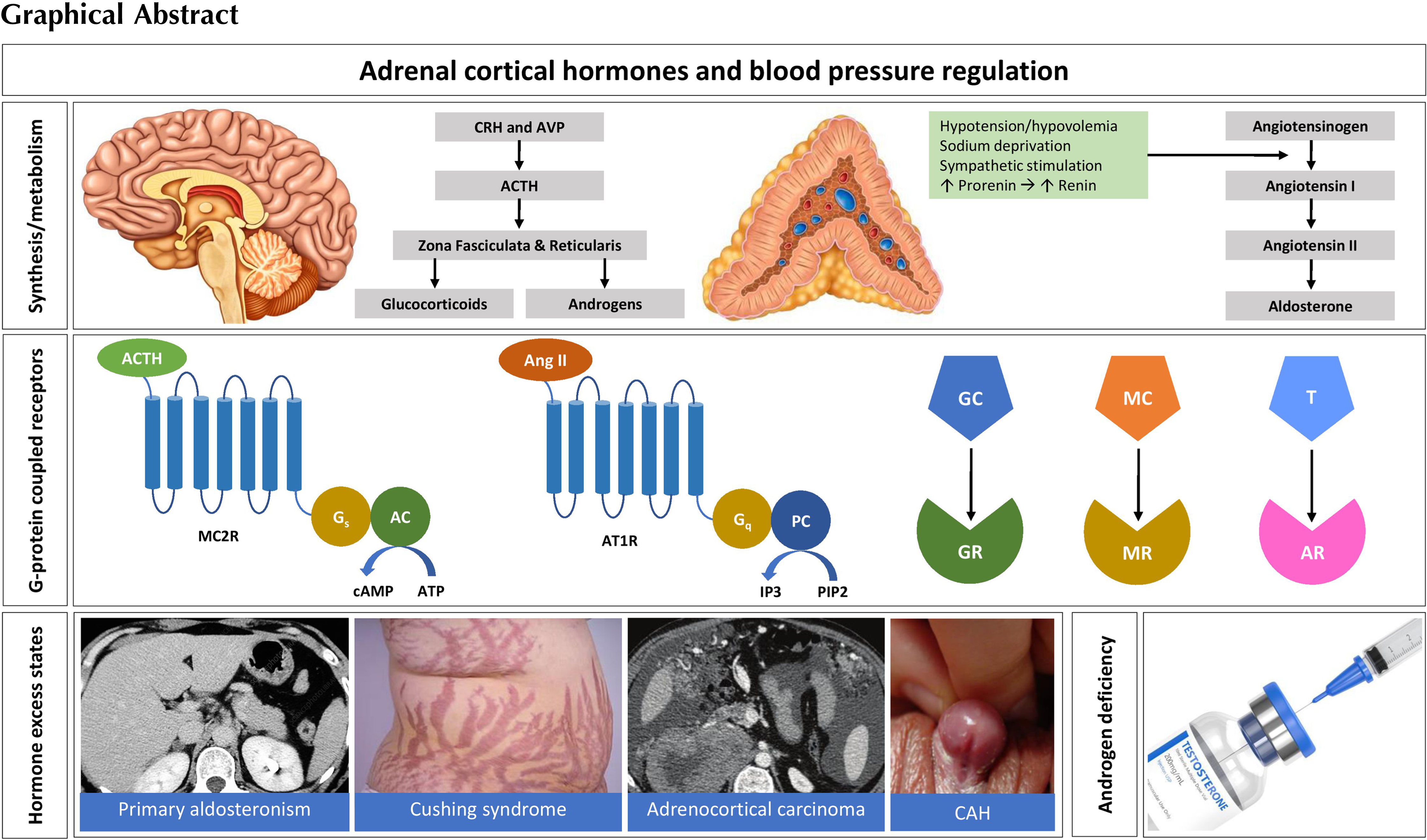
A depiction of the adrenal steroid synthesis pathways and their sites of synthesis is shown in Fig. 3.1 .
Regulation of steroidogenesis: cholesterol, StAR, and p450scc
Common to steroidogenic pathways is the initial conversion of cholesterol to pregnenolone by P450scc (cholesterol side chain cleavage enzyme, steroid 20–22 lyase, cholesterol 20–22 desmolase, or CYP11A1) that is present on the inner mitochondrial membrane. While adrenocortical cells are capable of de novo cholesterol synthesis, the majority comes from the uptake of LDL particles. Cholesterol esters are transferred to lysosomes where they are either converted into free cholesterol, or stored as cytosolic lipid particles to be released when required by hormone-sensitive lipase (HSL). Steroidogenic acute regulatory (StAR) protein facilitates the transfer of cholesterol across the mitochondrial membrane where P450scc resides. While not essential for steroidogenesis, the absence or dysfunction of StAR protein due to gene mutations results in severe and sometimes fatal hyposteroidaemia [ , ].
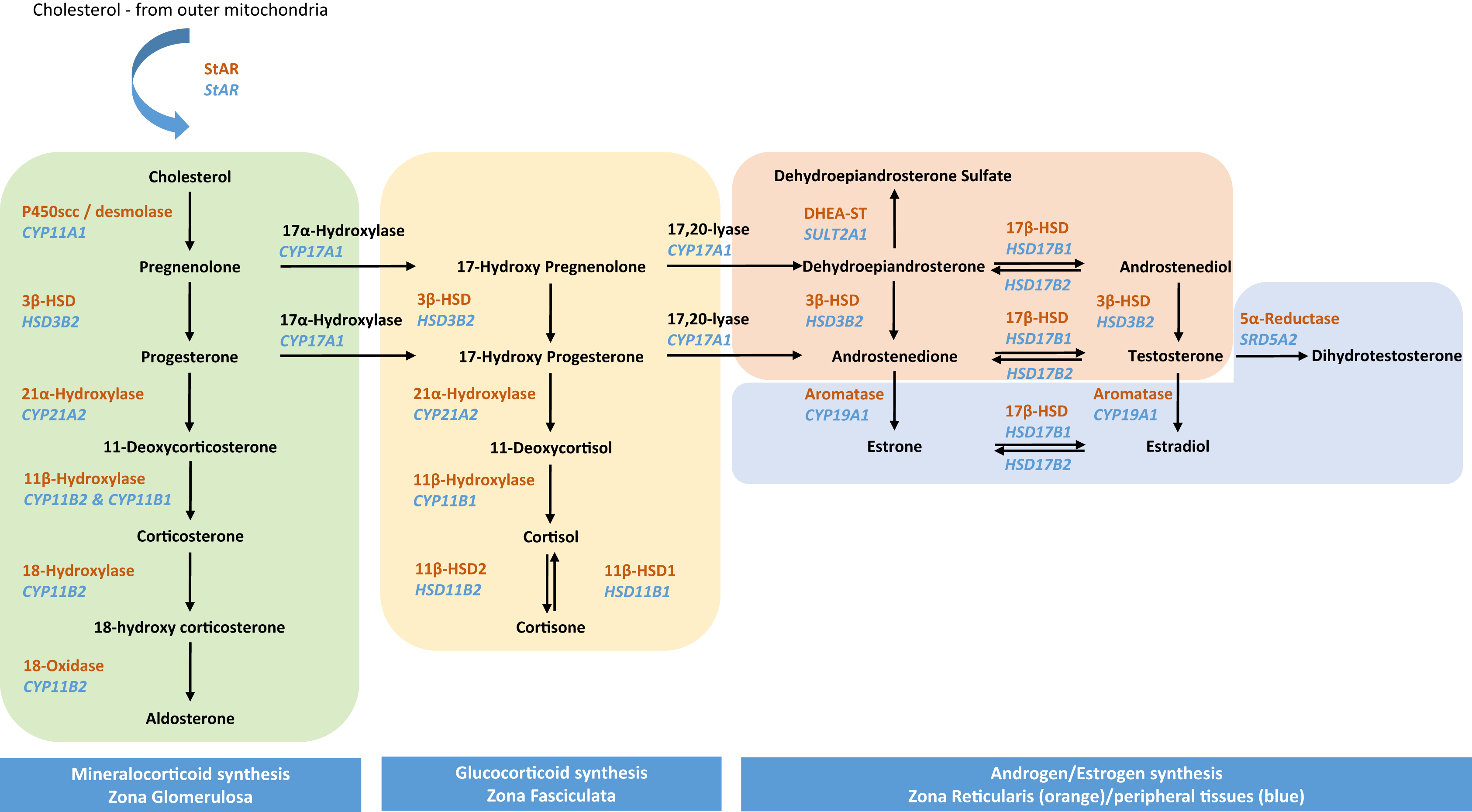
Adrenal steroidogenesis is regulated by rapid “nongenomic” cell signaling processes, as well as slower “genomic” processes that are dependent on gene expression. The predominant regulator of adrenal steroidogenesis is adrenocorticotropic hormone (adrenocorticotropin; ACTH) secreted from the anterior pituitary. ACTH binds to the transmembrane bound melanocortin type-2 receptor (MC2R), a G-protein-coupled receptor that exerts its effect through downstream cyclic adenosine monophosphate (cAMP). While the MC2R is predominantly found in the zona fasciculata, it is also located in the zona glomerulosa and to a lesser extent, the zona reticularis [ ]. ACTH regulates the activity and expression of various steroidogenic enzymes especially that of HSL and StAR protein which control levels of intracellular cholesterol and its transport within the mitochondrial matrix, respectively [ ]. This regulation occurs predominantly through cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathway, resulting in rapid phosphorylation of proteins involved in steroidogenesis, including HSL and StAR proteins (nongenomic response). In addition to the classical cAMP/PKA pathway, the cAMP can cross-talk with other signaling pathways such as mitogen-activated protein kinase/extracellular signal-regulated protein kinase (MEK/ERK) pathway to mediate phosphorylation of proteins involved in steroidogenesis [ ].
Simultaneously, ACTH also triggers a slower genomic response by stimulating the expression of various steroidogenic genes including StAR , CYP11A , MC2R , and melanocortin-2 receptor accessory protein which serve to upregulate the steroid synthesis. The half-life of StAR protein is very short, approximately 5 to 15 minutes. Hence, in addition to the rapid phosphorylation of steroidogenic enzymes, stimulation of expression of steroidogenic genes is important to replenish and maintain StAR protein expression. ACTH can directly activate the cAMP-responsive element binding protein (CREB), which is a key transcription factor involved in the regulation of Star transcription, through pulsatile phosphorylation. Additionally, ACTH can indirectly activate CREB through posttranscriptional modification of cotranscription factors [ ]. Fig. 3.2 shows the genomic and nongenomic pathways involved in the adrenal steroidogenesis under the influence of ACTH [ ].
Aldosterone synthesis in the zona glomerulosa
Aldosterone synthesis is usually confined to the zona glomerulosa. Its regulation and synthesis is dependent on the high levels of expression of angiotensin II type 1 receptors (AT1Rs) and aldosterone synthase (CYP11B2) found in this zone. CYP11B2 has both 11-hydroxylase and 18-hydroxylase activity and is responsible for the final stages of aldosterone synthesis ( Fig. 3.1 ). The other three steroidogenic enzymes required for aldosterone biosynthesis are P450scc, 3β-hydroxysteroid dehydrogenase (3β-HSD), and 21α-hydroxylase. These three enzymes are found in all adrenocortical zones; however, absent expression of CYP11B1 and CYP17A1 genes in the zona glomerulosa which encode 11β-hydroxylase and 17α-hydroxylase, respectively, prevents diversion of steroid synthesis toward cortisol or androgens [ ].
Aldosterone synthesis is stimulated predominantly by angiotensin II and plasma K + but is also acutely regulated by ACTH and locally produced endothelin-1 [ ]. Angiotensin-II acting via the AT1R and ACTH acting via the MC2R both lead to an increase in the availability of precursors, such as cholesterol and NADPH, the latter of which is an essential cofactor for CYP450 activity [ ]. Long-term upregulation of aldosterone synthesis involves the increased expression of StAR protein and aldosterone synthase [ , ].
In vitro studies indicate that angiotensin II binding to the AT1R induces activation of several signaling pathways in zona glomerulosa cells. Angiotensin II binding to the AT1R induces G-protein-mediated Ca 2+ signal generation and protein kinase C (PKC) activation, which activates diverse signaling pathways, including stimulation of MAPK, tyrosine kinases, lipoxygenase production, phospholipase D, and phospholipase A2 [ ]. Angiotensin II, however, also induces G protein-independent signaling pathways, such as β-arrestin-mediated activation of MAPK [ ]. Specifically, upregulation of aldosterone synthase involves Ca 2+ signal-mediated activation of calmodulin kinases (CAMKs), which regulates the transcription and/or activity of early transcription factors [ , ].
Like angiotensin II, K + shares calcium signaling as a key regulator of aldosterone production, with K + increasing intracellular calcium activating voltage-sensitive L- and T-type calcium channels resulting in the influx of calcium from extracellular sources [ ]. Adrenal zona glomerulosa cells are typically hyperpolarized by a predominant K + conductance facilitated by the “leak” K + channels of the 2-pore domain/transmembrane family, TASK1 (KCNK3) [ ]. Membrane polarity is also under the control of different Kir (K + inward rectifying) channels, many of which are G-protein coupled. An increase in K + functionally results in increased mobilization of intracellular calcium and the activation of the calcium-CAMK system signal cascade [ , ].
Although angiotensin II and K + regulate aldosterone production independently, they also act synergistically. When both angiotensin II and K + levels are low, they attenuate the effect of the other agonist on aldosterone production, whereas when the concentration of angiotensin II or K + is modest to high, small changes in the concentration of the other agonist elicit large increases in aldosterone production [ ]. Fig. 3.3 outlines the pathways involved in aldosterone synthesis regulated by angiotensin II [ ].

Cortisol synthesis in the zona fasciculata
The zona fasciculata does not express AT1R nor aldosterone synthase but expresses the MC2R and 11β-hydroxylase (CYP11B1), which, unlike CYP11B2, does not have 18-hydroxylase activity and thus cannot convert 18-hydroxycorticosterone to aldosterone and has minimal capacity to convert corticosterone to 18-hydroxycorticosterone [ ]. Both the zona glomerulosa and zona fasciculata express 21α-hydroxylase, but the expression of 17α-hydroxylase in the zona fasciculata selectively permits cortisol synthesis. Very little, if any, cytochrome b 5 is expressed in the zona fasciculata. Cytochrome b 5 is an important regulator of CYP17 side-chain cleavage activity and its presence is required for the efficient side chain cleavage activity of CYP17, but not for 17α-hydroxylase function. In consequence, the 17α-hydroxylation but not 17,20-lyase activity of CYP17 predominates in zona fasciculata resulting in the production of glucocorticoids (cortisol and corticosterone) but very little dehydroepiandrosterone (DHEA) [ ].
De novo biosynthesis of adrenal steroid hormones (adrenal steroidogenesis) is a dynamic process that is tightly regulated by multiple mechanisms including transcriptional and posttranslational modification of steroidogenic enzymes and proteins involved in the substrate transportation [ ]. In addition, various proteins involved in the cholesterol biosynthesis and uptake also function as regulatory factors of adrenal steroidogenesis. These regulatory mechanisms are extremely vital, as glucocorticoid hormones (cortisol in humans and corticosterone in rodents) are the key regulators of variety of physiological functions including blood pressure, inflammation, glucose metabolism, and stress. The endoplasmic reticulum-associated degradation (ERAD) is a mechanism to remove misfolded or unfolded proteins to maintain protein quality. Small vasolin-containing protein-interacting protein (SVIP) is an endogenous ERAD (endoplasmic reticulum associated protein degredation pathway) inhibitor. SVIP overexpression increases the cortisol and DHEA biosynthesis, whereas exaggerated overexpression led to apoptosis and diminished adrenal steroidogenesis. This indicates that SVIP is a novel regulator of adrenal cortisol and DHEA biosynthesis [ ].
Corticotrophin-releasing hormone (CRH) secreted by the paraventricular nucleus of the hypothalamus predominantly coordinates the central regulation of glucocorticoid secretion, potentiated by other hypothalamic products most importantly vasopressin [ ]. CRH activates CRH receptors on the corticotrophs of anterior pituitary to enhance ACTH secretion, which in turn stimulates glucocorticoid-secreting cells within the adrenal cortex signaling. The glucocorticoids send inhibitory feedback signs to the hypothalamic–pituitary–adrenal (HPA) axis, especially at the level of the anterior pituitary, but also at the level of the hippocampus and hypothalamus. The HPA axis has a characteristic dynamic ultradian rhythm manifested by oscillating levels of ACTH and glucocorticoid hormones, so that adrenal glands release glucocorticoids in near-hourly pulses. This ultradian rhythm becomes disrupted during aging and in stress-related disease such as major depression. The ultradian rhythm results from a sub-HPA system that functions as a peripheral hormone oscillator [ ].
Adrenal androgen synthesis and regulation in the zona reticularis
Unlike the zona glomerulosa and zona fasciculata, which replace the fetal adrenal zones early in the neonatal period, the zona reticularis develops slowly, becoming active in producing androgens at adrenarche at around 6–8 years [ ], and peaking in androgen production at around 30 years [ ]. During adrenarche, the gene expression profile of the zona reticularis changes leading to an increase in the synthesis of 17α-hydroxylase, cytochrome b 5, and P450 cytochrome oxidoreductase (POR) enzymes, and a decrease in synthesis of 3β-hydroxysteroid dehydrogenase type II (HSD3B2) [ ].
Cytochrome b 5 and POR form a quaternary complex with 17α-hydroxylase, which maximizes its 17–20 lyase activity. This pattern of expression of steroidogenic enzymes and cofactors in combination with the relatively low levels of 21α-hydroxylase and 11β-hydroxylase and the greater affinity of 17α-hydroxylase for pregnenolone and 17-hydroxypregnenolone over HSD3B2 enable DHEA synthetic pathways to predominate over those of cortisol synthesis in zona reticularis [ ].
DHEA is largely converted to its sulfated form, DHEAS, by the high levels of sulfotransferase 2A1 (SULT2A1) found in the zona reticularis [ ]. A small proportion of the DHEA produced is converted to androstenedione, some of which is converted into testosterone, presumably due to the presence of 17β-hydroxysteroid dehydrogenase-5 (HSD5) [ ]. The clinical significance, however, of these small quantities of adrenal testosterone is not established.
The regulation of adrenal androgen synthesis is predominantly dependent on the ACTH/cAMP/PKA and the MEK/ERK signaling pathways [ ]. Inhibition of MEK/ERK signaling affects CYP17 and HSD3B2 expression and thus androgen synthesis [ ].
In summary, the differential expression of enzymes in each zone of the adrenal cortex is responsible for zonal selectivity of the type of steroid synthesis and secretion.
Glucocorticoids, receptors, and cardiovascular system effects
Glucocorticoids and their actions via the glucocorticoid receptor (GR) have a role in the genesis and development of cardiovascular disease (CVD). Glucocorticoids are essential for life, coordinating a multitude of fundamental processes including metabolic homeostasis, immune and inflammatory responses, reproduction, development, cell proliferation, and cognitive function [ ]. Glucocorticoid actions are largely mediated through their binding to the GR, but newer studies indicating non-GR activity demonstrate a more complex interplay of corticosteroid action [ ]. The GR is evolutionarily related to the mineralocorticoid receptor (MR), another member of the nuclear receptor (NR) superfamily and steroid receptor subfamily that includes the progesterone receptor (PR), estrogen receptors (E2), and androgen receptors (ARs), and as such, there are emerging data showing cross-talk between steroid receptors and physiological and pathophysiological function .
The glucocorticoid receptor (GR)
The GR, like the other steroid hormone receptors, is a member of the NR superfamily, which share common features and mechanisms of action, and mainly act as ligand-inducible transcription factors that directly bind DNA and influence the expression of target genes. The GR is encoded by the NR3C1 gene located on chromosome 5q31-32 [ ]. Like other NRs, the GR consists of an N-terminal transactivation domain (NTD), a central DNA-binding domain (DBD), a C-terminal ligand-binding domain (LBD), and a linker domain connecting the DBD and LBD [ ]. The LBD contains the site for glucocorticoid binding as well as an activation function site (AF2) which undergoes a conformational change upon ligand binding enabling additional interaction with an array of coactivators or corepressors. The NTD binds a diverse range of coregulators and components of the transcriptional machinery through activation function domain 1 (AF1). It is the most varied region of gene and subject to greater polymorphism as a result of posttranslational modifications. The NTD cooperates synergistically with the LBD to reinforce the receptor structure [ , ]. Conversely, the DBD is a highly conserved two zinc finger motif that mediates dimerization and subsequent binding to specific genomic sequences called glucocorticoid response elements (GREs).
The GR exists in several isoforms generated by alternative splicing. To date, this includes GR-α (8 subisoforms), GR-β (8 subisoforms), GR-γ, GR-A, and GR-P. The GR-α isoforms have similar affinities for glucocorticoids and similar interaction abilities with GREs. In vitro studies, however, have demonstrated isoform specific differences in function [ ]. Unlike other GR isoforms, the GR-β isoform is located to the nucleus; it does not bind glucocorticoids and yet binds to GREs acting as a negative inhibitor of GR-α [ ]. Increased expression of GR-β has been associated with glucocorticoid resistance, which may be due to competition for GRE binding, competition for transcriptional co-regulators, or the formation of inactive isoform heterodimers. The ability of GR-β and other GRs to inhibit the activity of GR-α is suggestive of a key role in glucocorticoid resistance and insensitivity [ ]. The GR-γ, GR-A, and GR-P have low transactivation activities. It has been postulated that the tissue-specific and the individual differences in relative abundances of GR isoforms could be of significance in glucocorticoid action [ ].
G lucocorticoid receptor and blood pressure regulation
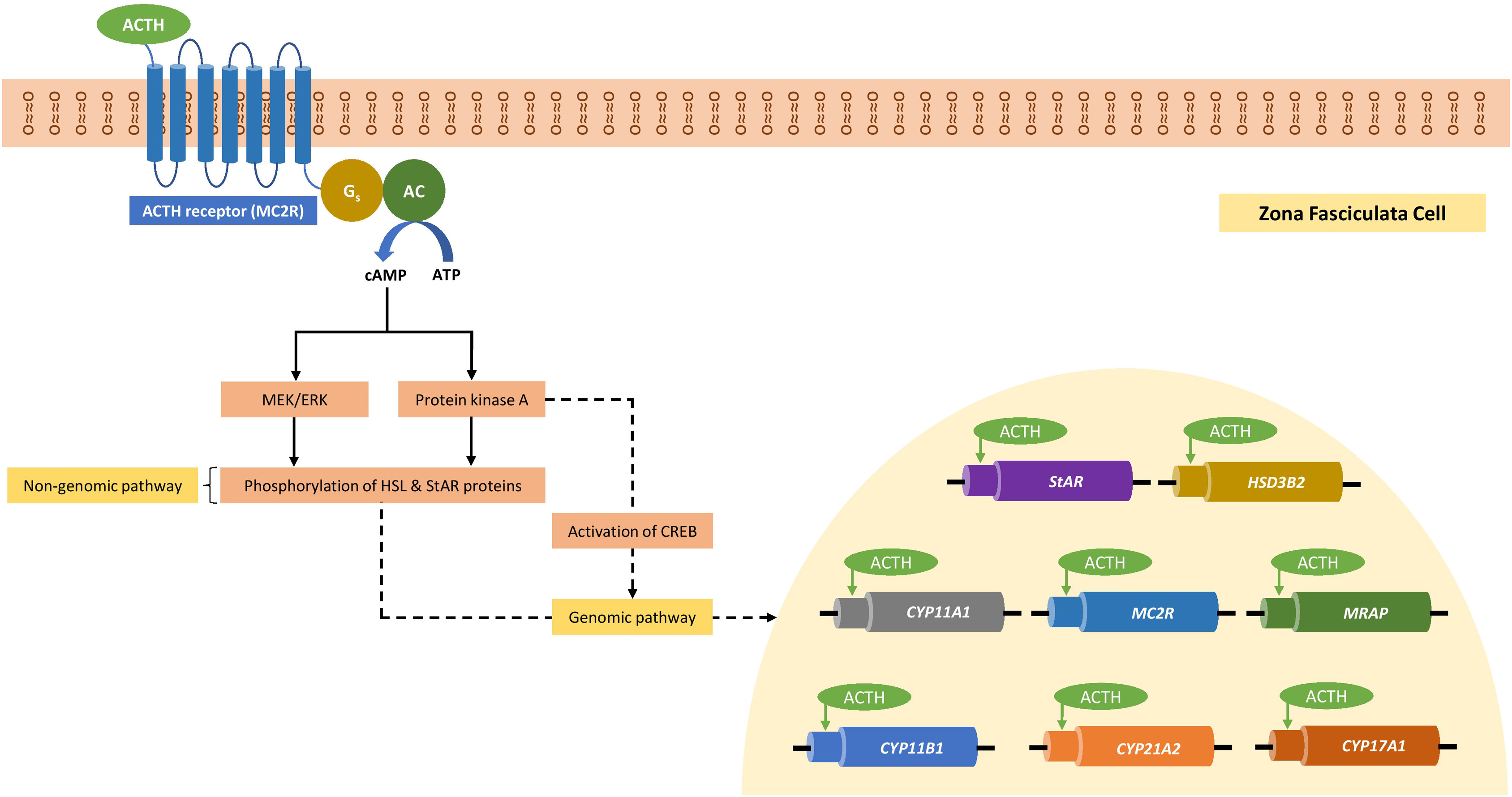
Blood pressure is dependent on the combined efforts of cardiac output and systemic vascular resistance and is regulated by baroreceptors. Hypertension is an established major risk factor for CVD. Glucocorticoids regulate the blood pressure and may cause hypertension [ ]. While it is traditionally believed that glucocorticoids influence blood pressure through activation of the MR, the presence of GRs in the vascular smooth muscle cells (VSMCs) and vascular endothelial cells (VECs) has shifted focus to the role of these tissue-specific receptors in modulating BP [ , ]. VSMC GR knock out (GRKO) mouse model studies demonstrate that while both control and GRKO mice had similar BP at baseline, the GRKO mice had attenuated acute and chronic hypertensive responses to dexamethasone in comparison to control mice suggesting a role for the GRs in VSMC in mediating the hypertensive response [ ]. Similarly, in a vascular endothelial GRKO mouse model, the GRKO mice had slightly elevated baseline BP but were moderately resistant to dexamethasone-induced hypertension compared to control mice, indicating the important role of GRs in VECs in BP regulation [ ].
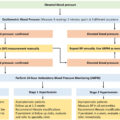
In cell culture experiments, glucocorticoids increase the expression of angiotensin II receptor type I (AT-1) in VSMCs, which have been postulated as a mechanism for increasing BP [ , ]. Studies have also demonstrated a role for both the GR and the MR in BP regulation by mediating the influx of Ca 2+ and/or Na + into VSMCs [ ]. Cell culture studies have also indicated that glucocorticoids may regulate BP through alternative pathways in VECs. One such study demonstrated that through nongenomic GR cell signaling, glucocorticoids influence generation of nitric oxide (NO) in VECs via nitric oxide synthase (NOS) activity, which induces vasodilation in VSMCs as well as acts locally to prevent platelet and leukocyte aggregation and inhibit VSMC proliferation [ ]. Fig. 3.4 shows graphical representation of the role of glucocorticoids and GR in the regulation of human blood pressure [ , ].
Mineralocorticoids, receptors, and effects on the cardiovascular system
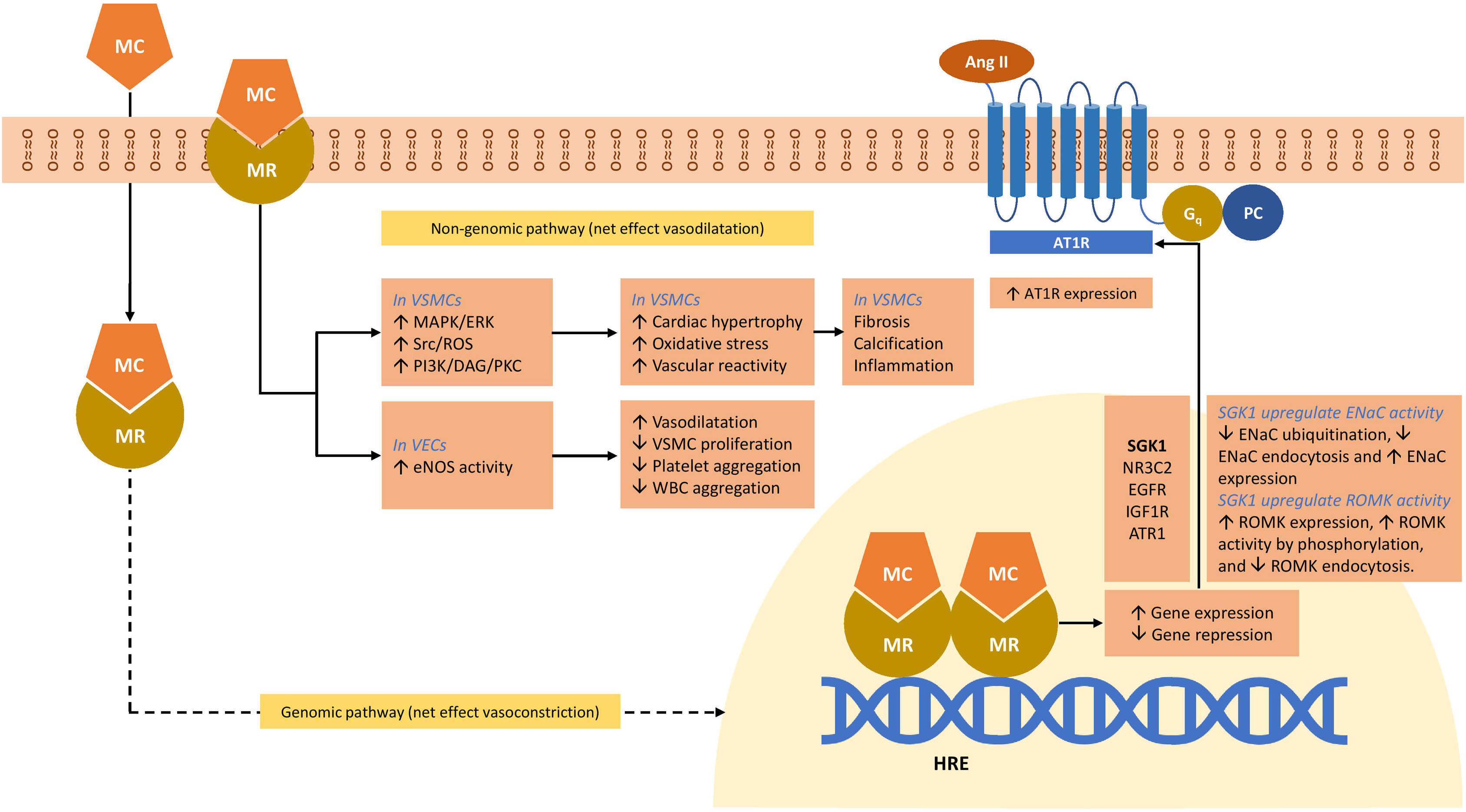
The primary function of aldosterone, the main mineralocorticoid, is to increase sodium reabsorption, and subsequent water reuptake in the kidney, thus maintaining normal blood pressure. The classic understanding of how aldosterone mediates its effect is through binding to the MR, which acts as a transcription factor for responsive genes, including that of the epithelial sodium channel, ENaC. Increased number of ENaC channels in the apical membrane results in enhanced reabsorption of sodium, which in turn promotes water reuptake [ ].
Unlike other steroids with cross-(steroid) receptor affinity, aldosterone binds only to the MR. Interestingly, the emergence of the MR preexisted aldosterone synthase, which coincided with the emergence of land vertebrates, and therefore, it is presumed that other steroid ligands are likely to have physiological effects [ ]. In fact, it is long established that 11-deoxycortisol and cortisol also act as MR agonists and have similar affinities to the MR as that of aldosterone. However, aldosterone is still the primary ligand for the MR in epithelial tissues at least, despite the significantly more abundant cortisol. This is due to the presence of colocalized 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) that converts cortisol to the inactive cortisone [ ].
The MR is widely distributed in a range of epithelial and nonepithelial tissues [ ], including the kidney, the cardiovascular system, the central nervous system, the gonads, and the colon [ ]. The function of the MR in many of these tissues is not fully elucidated, but in tissues where there is an absence of colocalized 11βHSD2, it is suggested that the MR acts as an additional or alternative GR [ ].
The mineralocorticoid receptor
The human MR (encoded by NR3C2 gene) is the longest of the NRs, containing 984 amino acids, and like the other members of the NR superfamily, it is comprised of three main functional domains: an N-terminal domain (NTD), a highly conserved DBD, and an LBD [ , ]. The NTD is intrinsically disordered allowing structural flexibility and diverse protein interactions [ ]. Like the GR, the MR contains an activation function (AF-1) region that is refuted to be able to bind a wide array of corepressors [ ].
When unbound to a ligand, the MR is localized to the cytoplasm where it is complexed with several chaperone proteins [ , ] which play a dynamic role in determining ligand-affinity [ ]. The LBD interacts with chaperone proteins which retain the inactive MR in the cytoplasm, in a high affinity state [ ]. Ligand binding induces a conformational change in the LBD, which (1) releases the chaperones enabling translocation to the nucleus and (2) creates the activation function-2 region (AF-2) which is a site for binding of transcriptional coregulators. In the nucleus, the dimerized MR–ligand complex recruits coregulators and transcription machinery and binds to hormone response element (HRE) in the promoter of MR-regulated genes [ ].
MR and blood pressure regulation: genomic effects
In kidney epithelial cells, MR activation upregulates the expression of proteins important in electrolyte homeostasis including ENaC, Na + /K + ATPase, renal outer medullary potassium channel, and serum- and glucocorticoid-inducible protein kinase 1 (SGK-1) [ ]. SGK-1 is a signaling molecule, upregulated as early as 30 min after the MR activation whose synthesis is related to the increased cell surface density of ENaC [ ]. SGK-1 inhibits the ENaC degradation [ ]. Coexpression of SGK-1 and ENaC increases the sodium channel activity by fourfold [ ].
SGK-1 is upregulated in several fibrotic diseases including myocardial fibrosis, lung fibrosis, liver cirrhosis, diabetic nephropathy, and glomerulonephritis [ ]. In the myocardium, SGK-1 promotes myocardial fibrosis by increasing fibrotic mediators such as connective tissue growth factor and transforming growth factor-β (TGF-β); proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β); and NADPH oxidase [ ].
Apart from mineralocorticoids, SGK-1 is upregulated by a variety of hormones including glucocorticoids (GR activation), 1,25-dihydroxyvitamin D3, gonadotropins, and TGFβ [ ]. Cardiomyocytes have both glucocorticoid and mineralocorticoid receptors. Glucocorticoid receptor agonist dexamethasone activates both GRs and MRs to upregulate SGK-1, which in turn activates a wide variety of transporters including Na + /H + exchanger 1 (Nhe1), which is a key factor in cardiac remodeling [ ].
In VSMC studies, aldosterone has been demonstrated to stimulate the expression of genes involved in fibrosis, calcification, and inflammation [ , ].
MR and blood pressure: non – genomic actions
Beyond these classic genomic effects, aldosterone also stimulates other, more rapid molecular pathways, which can be MR-independent or involve cross-talk between MR and other receptors [ ]. In renal epithelia, aldosterone has been shown to cause tyrosine kinase c-Src-mediated transactivation of the epidermal growth factor receptor and the subsequent activation of the extracellular signal-regulated kinase 1/2 (ERK 1/2) and a rapid and transient increase in intracellular Ca 2+ [ , ].
In VSMCs, stimulation with aldosterone has been shown to cause an increase in phosphorylation of tyrosine kinase c-Src via both G-protein-dependent and independent pathways, leading to subsequent activation of ERK 1/2, MAPK, and NADPH oxidase pathways resulting in profibrotic and proinflammatory pathways [ ]. Interestingly, ERK activation can be blocked by an AT1R blocker [ ], and several reports [ , ], in disparate cell lines, have demonstrated cross-talk between these two receptors: MR and AT1R. In cardiomyocytes, aldosterone also stimulates apoptosis via the activation of ERK 1/2 and mitochondrial superoxide formation [ ]. Activation of PI3K pathways leads to increased endothelial nitric oxide synthase activity in VECs, and increased contractility of VSMCs [ ]. Fig. 3.5 depicts the role of mineralocorticoids and MR in the blood pressure regulation [ , ].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


