A Brief Overview of Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is understood to be the product of genetic and epigenetic changes within a multipotent myeloid stem/progenitor cell that generate and amplify a clone capable of differentiation arrest and uncontrolled proliferation. This clone, completely unmoored from the genetic machinery and stromal signaling that may normally regulate its maturation and expansion, propagates unceasingly, suffocating the bone marrow and compromising the production of functional blood cells. AML may spring seemingly de novo from a previously silent and newly activated clone or may represent the evolution (via progressive clonal mutagenesis) of preexisting neoplasms such as a myelodysplastic syndrome (MDS) or a myeloproliferative neoplasm (MPN). The origin of these pathologic mutations remains unknown in most cases, although AML may be associated with a number of environmental factors in rare instances (industrial chemicals, tobacco, radiation, chemotherapy).
In the United States, it is estimated that there are more than 20,000 new cases of AML per year, a number that seems to be gradually increasing for unclear reasons. AML is diagnosed via bone marrow aspirate and biopsy wherein blast forms must account for at least 20% of the biopsy cellularity (or alternatively 20% of the peripheral blood white cell count). Prognostic and treatment decisions are made on the basis of the cytogenetic and molecular genetic characteristics of the malignant clone, with consideration given to the age, comorbidities, and functional status of the patient.
AML is swiftly and universally lethal if untreated and requires timely initiation of cytotoxic induction chemotherapy, with the goal to induce remission. Anemia, infection, and bleeding are nearly ubiquitous during the early phases of treatment. If remission is achieved, decisions regarding appropriate consolidation therapy depend on patient age and comorbidities, as well as the cytogenetic and genomic characteristics of the leukemia. Younger, fitter patients or those with higher-risk disease may be offered allogeneic hematopoietic stem cell transplantation (ASCT), whereas older patients, those with prohibitive comorbidities, or those with lower-risk disease may proceed to consolidation chemotherapy. Fewer than one-third of patients survive 5 years after diagnosis, with relatively better outcomes among those who are younger and fitter, have lower-risk disease, and have access to suitable hematopoietic stem cell donors.
Basic Pathophysiologic Principles
Blood cells of the myeloid lineage are those that trace their development to a common myeloid progenitor, a multipotent hematopoietic stem cell which, under the influence of medullary stromal growth factors, gives rise to all nonlymphoid blood cells (including granulocytes, erythrocytes, and platelets). AML is a malignancy of myeloid precursor cells. It is the result of impaired maturation and unrestrained proliferation leading to the accumulation of immature nonfunctional myeloid cells.
The pathogenesis of AML is a clonal process born of a somatic mutation within a single hematopoietic stem/progenitor, and potentiated by the accrual of additional dysregulating mutations among that cell’s progeny. Along the pathway from sentinel mutation to acute leukemia, this clonal evolution may take on a series of intermediate phenotypes in the form of premalignant proliferative disorders. These “intermediate phenotypes” form a spectrum ranging from seemingly dormant clones detectable only by genomic studies (clonal hematopoiesis of indeterminate potential [CHIP]), to relatively benign cytopenias of clonal origin (clonal cytopenias of undetermined significance [CCUS]), to clinically significant malignant states such as the myeloproliferative neoplasms (MPNs) and MDSs. Although the transformation from a healthy patient to a leukemic one may often seem abrupt, or acute, it in fact occurs in a series of intermediate steps all culminating in the generation of an aggressively malignant clone. Sometimes these intermediate stages are quite discrete and evident, as is the case when a chronic MPN or MDS undergoes leukemic transformation. In other instances, the clonal state preceding AML may be quiescent until the time of transformation. Adding to the complexity of leukemic pathogenesis is the critical observation that not all acquired mutations are destined to beget leukemia; in fact, only a minority of patients with MPN or MDS ever experience transformation, and only a minority of patients with CHIP or CCUS progress to development of clinically evident disease.
The specific sequences of genomic events that generate leukemic clones are complex and involve both genomic and epi-genomic alterations that lead to clonal expansion, accumulation of undifferentiated myeloid cells, and compromise of normal hematopoiesis.
Epidemiology
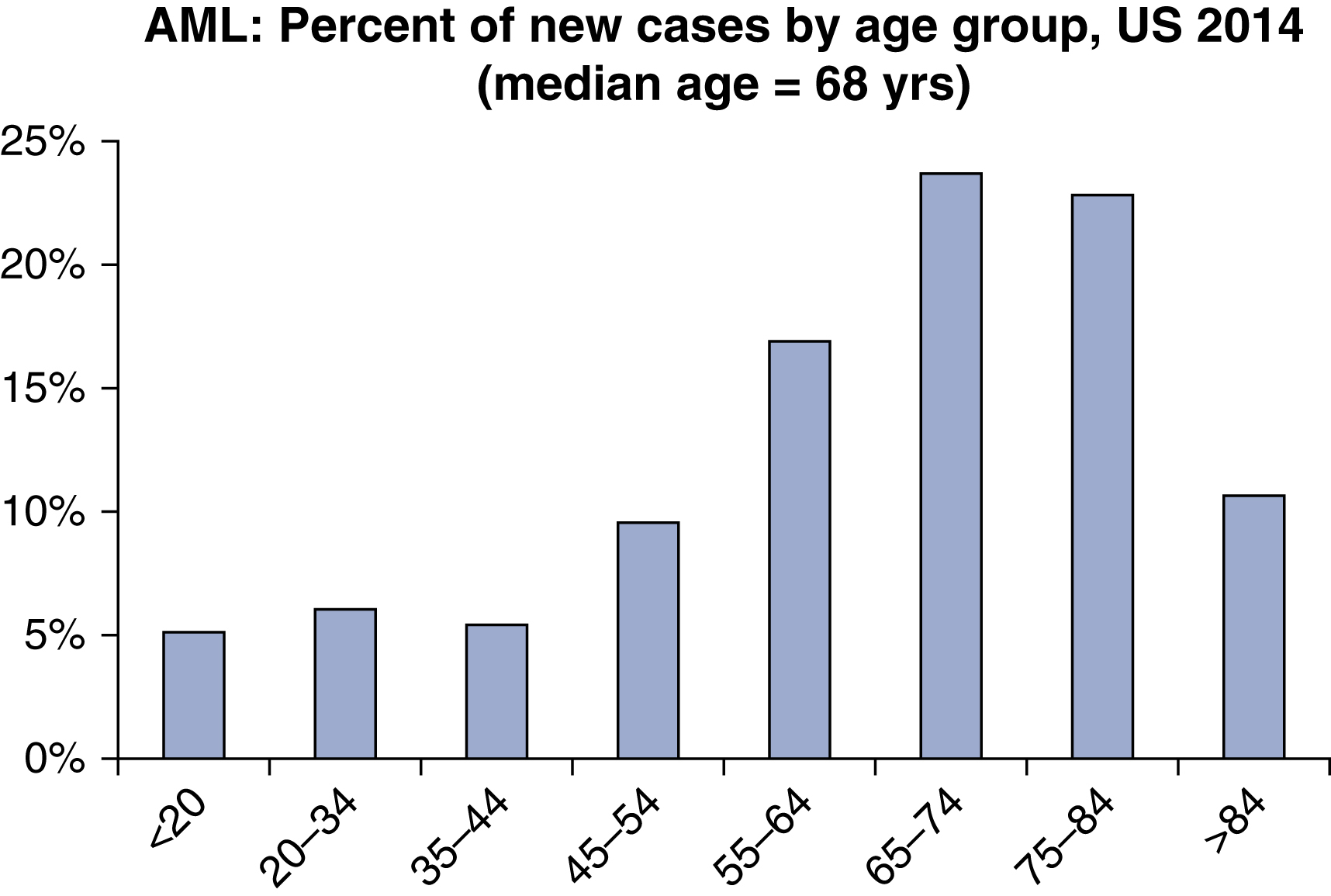
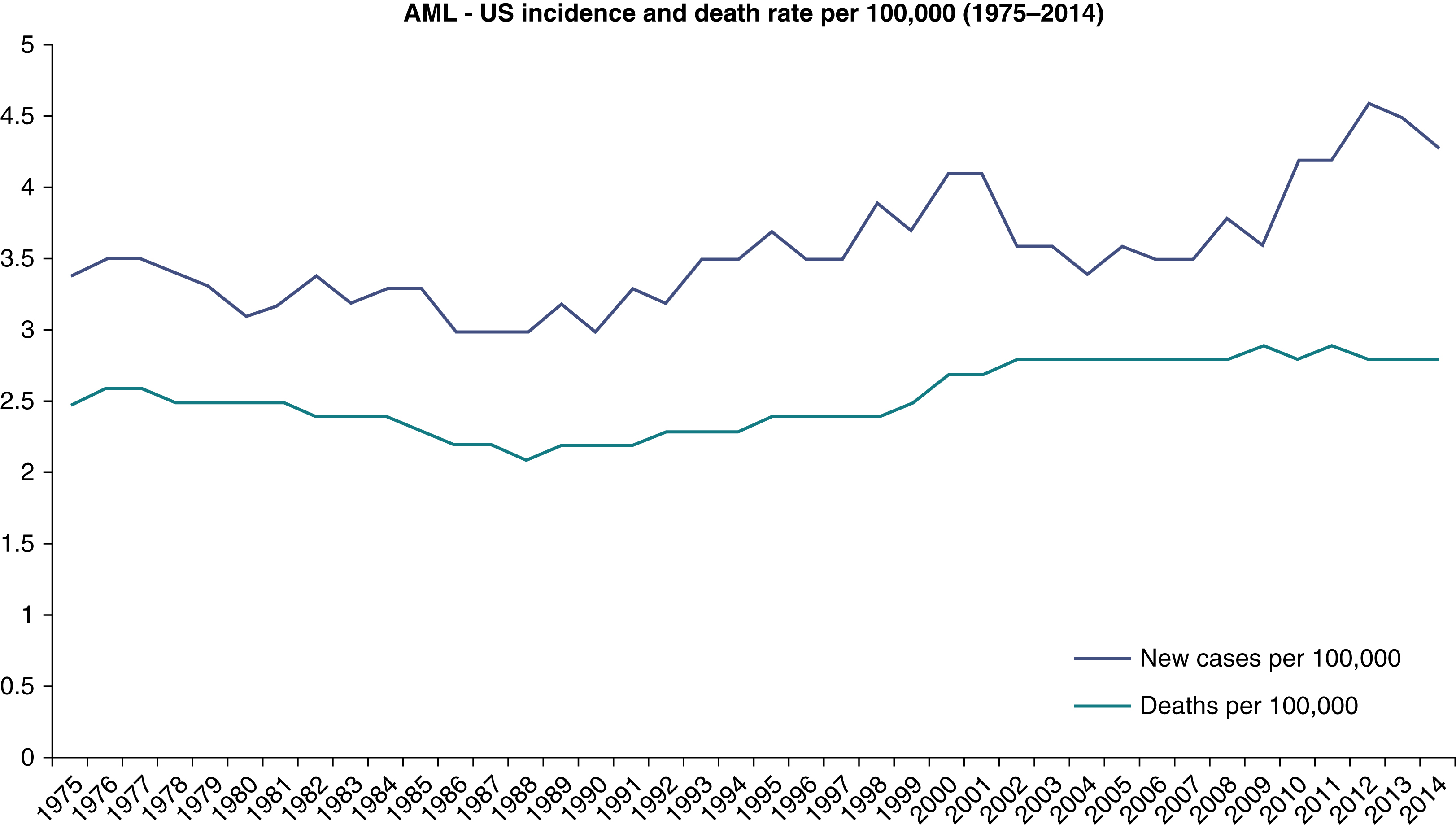
AML is a relatively rare disease. It accounts for only 1.3% of all new cancer cases in the United States. However, it is the most common acute leukemia among adults, accounting for approximately 80% of such cases. The rate of incidence in the United States has been reported to be 4.2 new cases per 100,000 people per year. The lifetime risk of acquiring AML in the United States has been estimated to be 0.5%. In 2017, an estimated 21,380 people were diagnosed with AML in the United States, and an estimated 10,590 died of the disease. It is more common among men than woman (incidence ratio 5.2:2.6). It may be somewhat more common among white individuals compared with other races. AML may be considered a disease of aging, with a median age at diagnosis of 68 years, and more than half of cases occur in patients older than 65 years ( Fig. 8.1 ). However, there is considerable heterogeneity of age, with nearly a quarter of cases occurring before age 50 years. The incidence of AML has been rising at a rate of 3.1% per year over the past decade for unclear reasons ( Fig. 8.2 ). However, the annual death rate has remained stable due to steady improvement in the 5-year survival rate, which is now approximately 27%. Survival rates are significantly better for younger and fitter patients, those with fewer medical comorbidities, those with cytogenetically favorable disease, and those with suitable hematopoietic stem cell donors.
Most cases of AML are deemed to be sporadic, although rare cases are associated with known germline genetic disorders (such as Fanconi anemia, trisomy 21, or uncommon familial mutations in genes such as CEBPA or RUNX1 among others). Preexisting clonal disorders of the myeloid lineage such as chronic myeloid leukemia (CML), MDS, MPNs, or more rarely aplastic anemia (AA) and paroxysmal nocturnal hemoglobinuria (PNH), are well-established risk factors for AML. Rates of leukemic transformation vary across and within the conditions and are often dependent on the specific genetic characteristics of each case.
Clinical Presentation
The most common presenting symptoms among patients with AML tend to be manifestations of pancytopenia ( Table 8.1 ). Rarely, hyperleukocytosis due to severely elevated blast count may precipitate leukostasis or microvascular compromise owing to obstruction by leukemic blasts.
| Anemia | Fatigue, Weakness, Pallor |
|---|---|
| Thrombocytopenia | Petechiae, ecchymosis, epistaxis, gingival bleeding, menorrhagia, retinal or vitreous hemorrhage |
| Neutropenia | Active infection Febrile neutropenia |
| Hyperleukocytosis | Pulmonary, neurologic symptoms, dyspnea, hypoxia, visual changes, headache, dizziness, confusion, somnolence |
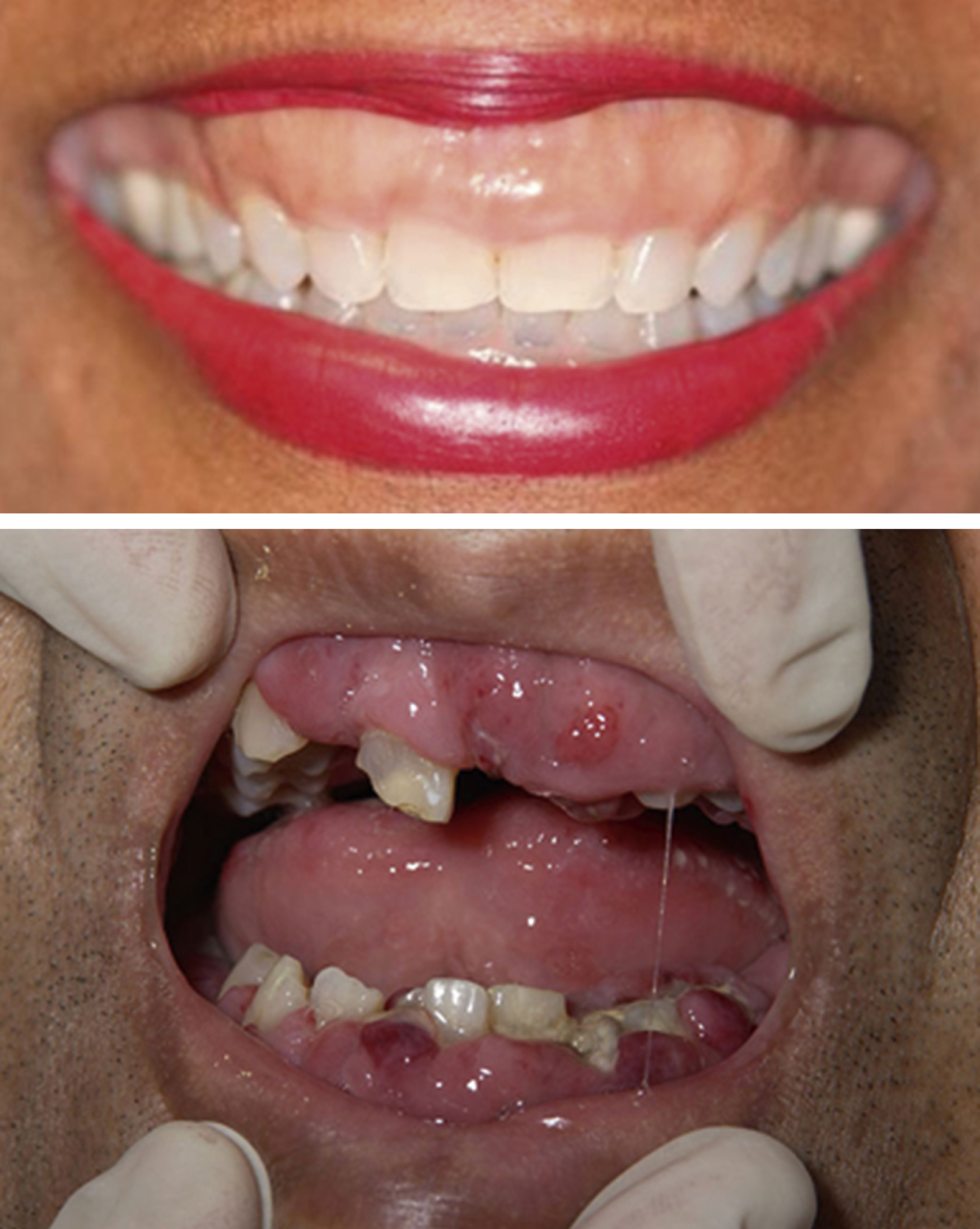
| Extramedullary disease | Gingivae ( Fig. 8.3 ) Skin-leukemia cutis ( Fig. 8.4 ) Chloromas/myeloid sarcoma—large bulky deposits of myeloid blasts at any anatomic location |
| Central nervous system involvement | Rare. Can present with headache, nausea, cranial nerve palsies, visual changes, gait disturbance |
Extramedullary disease is not uncommon and may occur to varying degrees in more than 20% of cases, whether concomitant with or independent of bone marrow involvement. The presence of extramedullary disease is not an independent prognostic factor and does not indicate higher-risk disease.
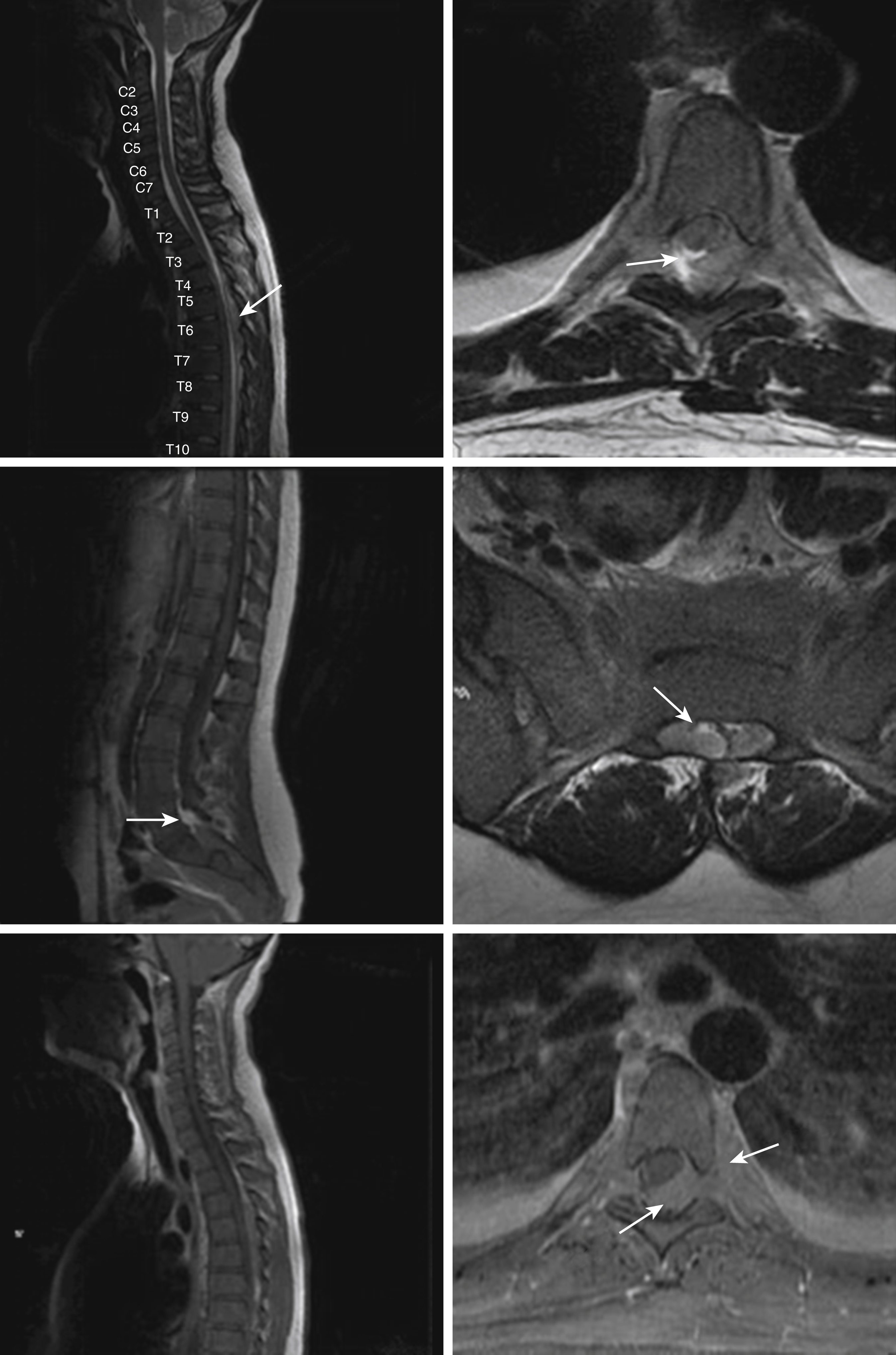
Central nervous system (CNS) involvement is relatively rare compared with lymphoblastic leukemias and aggressive lymphomas. The exact incidence is unknown as it is not routinely tested for in the absence of symptoms. CNS disease may often be asymptomatic. A CNS chloroma may present with symptoms of mass effect such as headache and nausea if present intracranially or as back pain or radicular symptoms if present in the spine. CNS involvement is, however, more often leptomeningeal, with manifestations including cranial nerve or peripheral nerve palsies, visual changes, and gait disturbances or sensations of imbalance ( Fig. 8.3 ).
Case 1
A 68-year-old man with a history of chronic alcohol dependence, scleroderma, and long-term immunosuppression presents with several weeks of progressive fatigue and weakness. He is found to be febrile and tachycardic. The examination is notable for pallor and oral petechiae. Laboratory evaluation is notable for pancytopenia. Initially, the admitting physicians consider the possibility that the patient’s pancytopenia might be due to chronic alcoholism, use of high-dose immunosuppressive medications, or acute infection. However, suspicion for a primary hematologic process remains high and seems to be confirmed when many myeloid blasts are observed on the patient’s blood smear.
Case 2
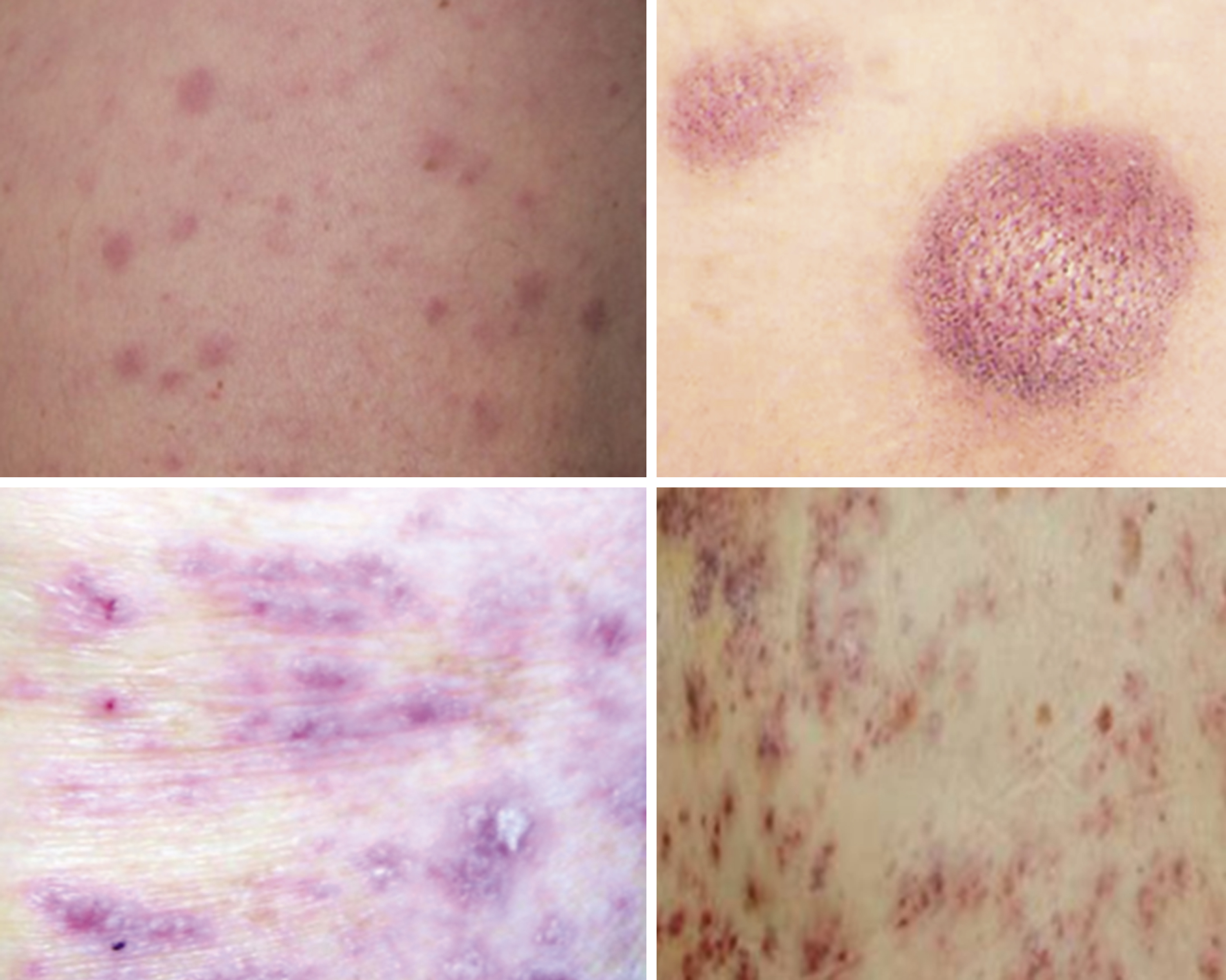
A 39-year-old woman with no past medical history presents with several weeks of fatigue progressing to dyspnea, somnolence, and confusion in recent days. She is afebrile, mildly hypertensive, and slightly hypoxic. The examination is notable for decreased sensorium, dried blood at the nares and mouth, somewhat hypertrophic gums (as in the upper portion of Fig. 8.4 ), decreased breath sounds bilaterally, and a diffuse nodular violaceous rash on her back and lower extremities (as in the upper left portion of Fig. 8.5 ). Laboratory evaluation demonstrates anemia, thrombocytopenia, and profound leukocytosis in excess of 200 × 10 9 /L. Blood smear reveals that the vast majority of these white cells are monoblasts. The patient’s neurologic and pulmonary signs and symptoms are deemed likely due to leukostasis, and her gum and skin findings are deemed likely manifestations of extramedullary disease (gingival hyperplasia and leukemia cutis, respectively).
Laboratory Manifestations
The most prominent laboratory evidence of AML is found in the complete blood cell count (CBC). Anemia and thrombocytopenia, owing to the loss of functional hematopoietic tissue to rapidly proliferating leukemic blasts, are nearly omnipresent, albeit to varying degrees ( Table 8.2 ). In the setting of hyperproliferative disease, with high peripheral blast count, a metabolic panel may show evidence of tumor lysis syndrome (TLS). Coagulation parameters may show evidence of disseminated intravascular coagulation (DIC), including elevations of the prothrombin time (PT), activated partial thromboplastin time (APTT), and D-dimer, as well as hypofibrinogenemia. DIC may be especially severe in APL.
Case 3
A 65-year-old woman with a history of hypertension and chronic kidney disease (stage 3) presents with a few days of subjective fevers, chills, and a productive cough. She is indeed febrile on presentation as well as tachycardic, with examination notable for crackles at the left lower lung field. Chest x-ray confirms an infiltrate at the site of abnormal auscultation. Her CBC is notable for new anemia and thrombocytopenia, as well as leukocytosis to 45 × 10 9 /L ( Fig. 8.6 ). Automated differential notes a near absence of mature neutrophils. Manual review of the blood smear demonstrates a preponderance of likely myeloid blast forms. A complete metabolic panel (CMP) demonstrates acute kidney injury with creatinine doubled from baseline, hyperkalemia, hyperphosphatemia, hypocalcemia, hyperuricemia, and elevated lactate dehydrogenase (LDH) ( Box 8.1 ). This patient is deemed to have likely AML, complicated by pneumonia in the setting of neutropenia, as well as by TLS (as evidenced by the abnormalities noted on the CMP). TLS is more likely in hyperproliferative presentations (those with high presenting blast counts) and among patients with baseline renal dysfunction, as in this case. In general, TLS is more likely to occur after initiation of chemotherapy rather than at presentation. Prompt identification and treatment is of paramount importance as chronic renal failure may result if the process goes unchecked.
Sodium: 145 mEq/L (135–145)
Potassium 7.1 mEq/L (3.5–5.0)
Chlorine: 98 mEq/L (96–106)
Carbon dioxide: 17 mEq/L (23–29)
Blood urea nitrogen: 29 mg/dL (7–20)
Creatinine: 3.9 mg/dL (0.5–1.2)
Calcium: 7.8 mg/dL (8.5–10.2)
Phosphorus: 6.3 mg/dL (2.4–4.5)
Urate: 9.7 mg/dL (2.4–6.0)
Lactate dehydrogenase: 1,097 U/L (160–450)
Case 4
A 71-year-old man with no past medical history presents with several days of persistent epistaxis. Review of systems is notable for recent malaise and low energy level as well as copious gum bleeding during tooth brushing. He is borderline hypotensive and mildly tachycardic. The examination reveals pallor, evidence of recent nosebleed, and oral petechiae. He has persistent oozing after venipuncture that resolves only after several minutes of forceful tamponade. The CBC with differential demonstrates anemia and thrombocytopenia with leukocytosis to 45 × 10 9 /L with 70% monocytes. Further laboratory evaluation demonstrates elevated international normalized ratio (INR), PTT, and D-dimer, as well as hypofibrinogenemia ( Box 8.2 ). This patient is deemed to have likely AML with monocytic differentiation (AML M5) manifesting with DIC. Although DIC is most common and tends to be most severe in APL, it is also common in monocytic and monoblastic variants of AML. Prompt recognition and treatment is paramount for the prevention of potentially catastrophic bleeding.
Platelets: 21 × 10 9 /L (150–450)
International normalized ratio (INR): 1.9 (<1.2)
Partial thromboplastic time: 44 seconds (25–35)
D-dimer: 19.3 mg/L (<0.5)
Fibrinogen: 91 mf/dL (150–400)
| Anemia, thrombocytopenia, neutropenia, leukocytosis, peripheral blood blasts |
| Tumor lysis syndrome: elevated serum urate, phosphate, potassium, lactate dehydrogenase, creatinine; low serum calcium |
| Disseminated intravascular coagulation: elevation of PT, APTT, D-dimer hypofibrinogenemia |
Pathologic Features
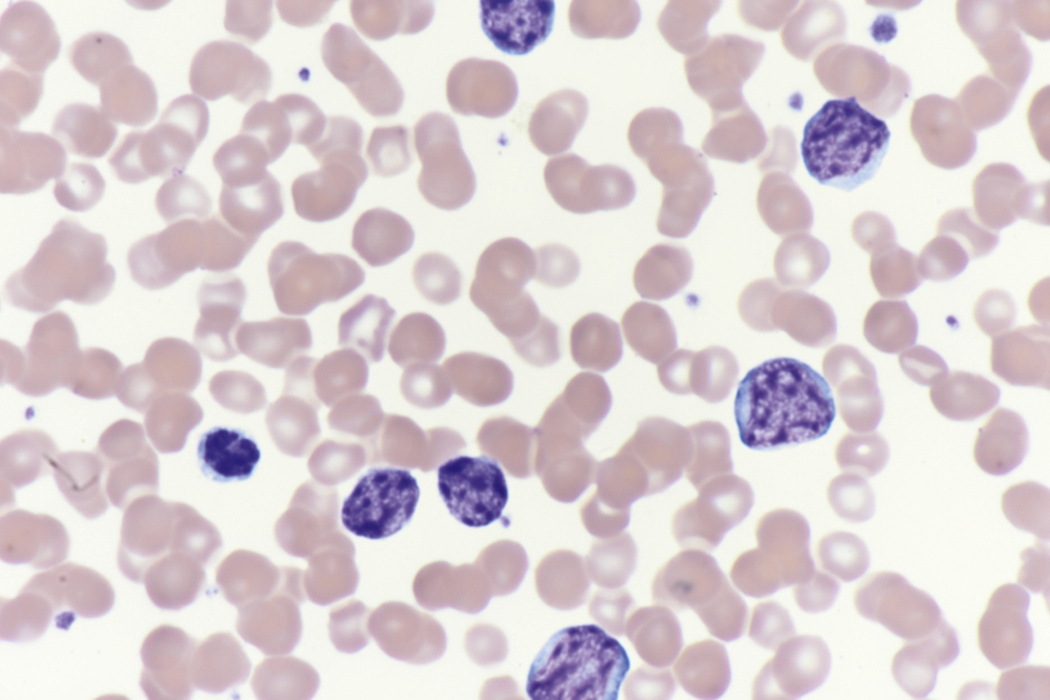
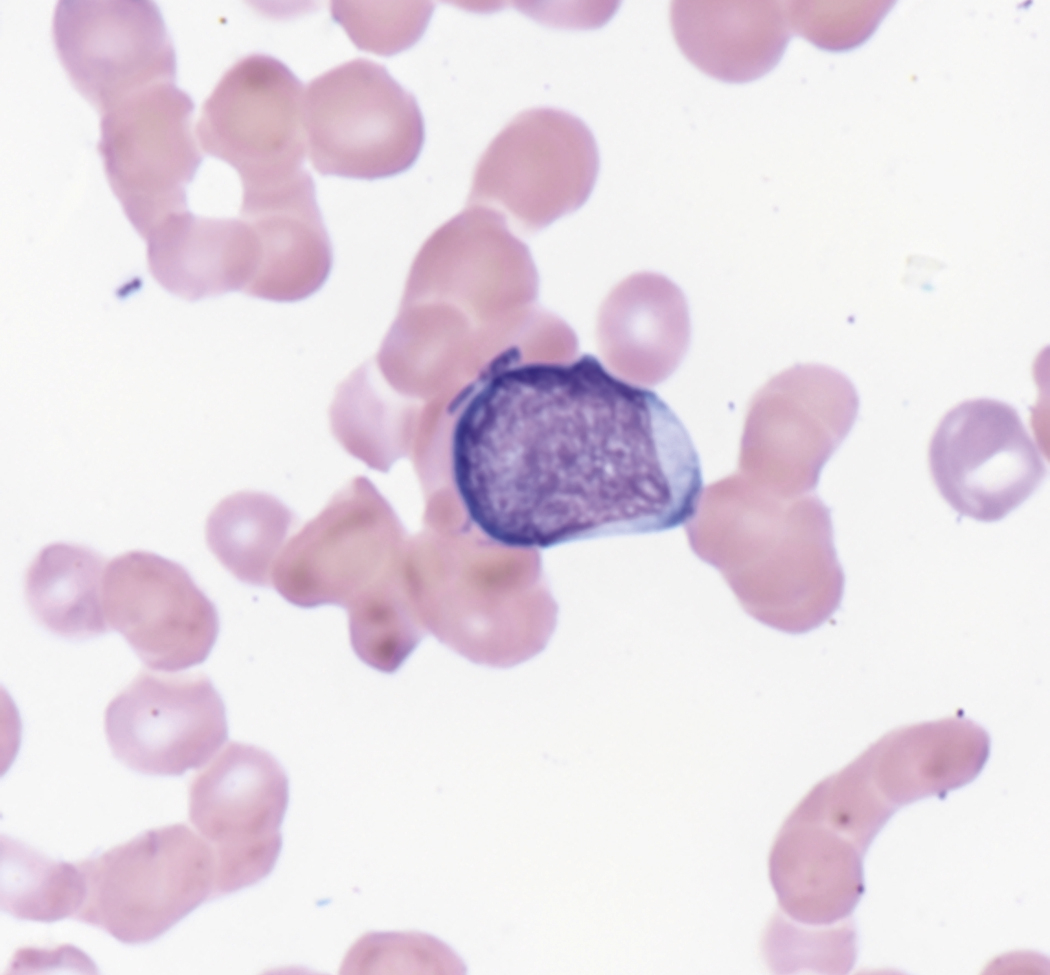
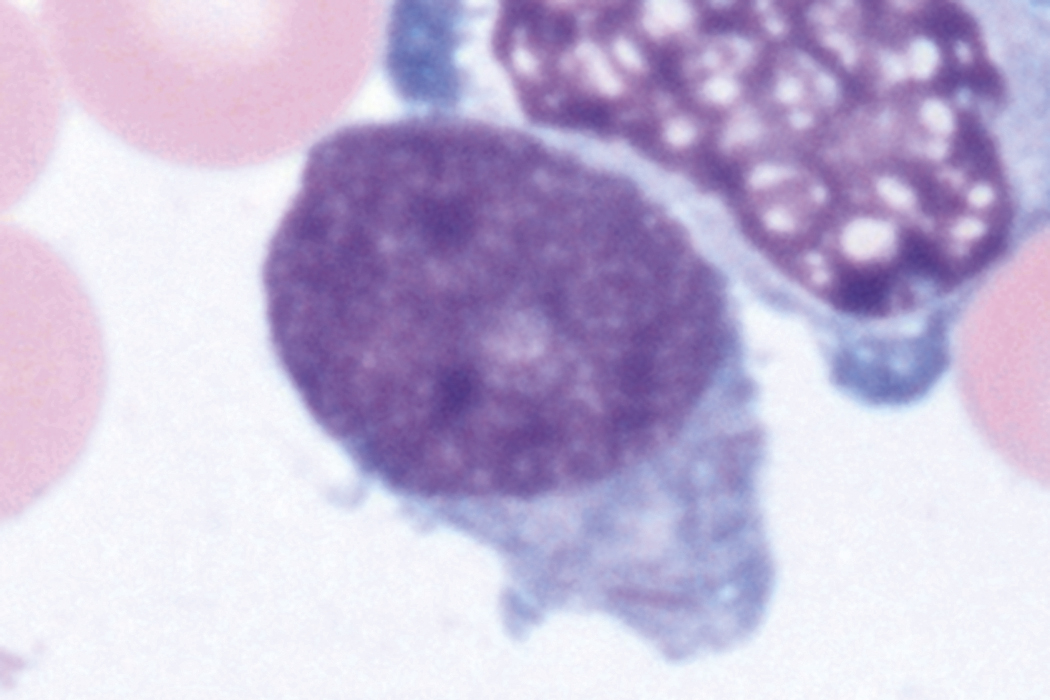
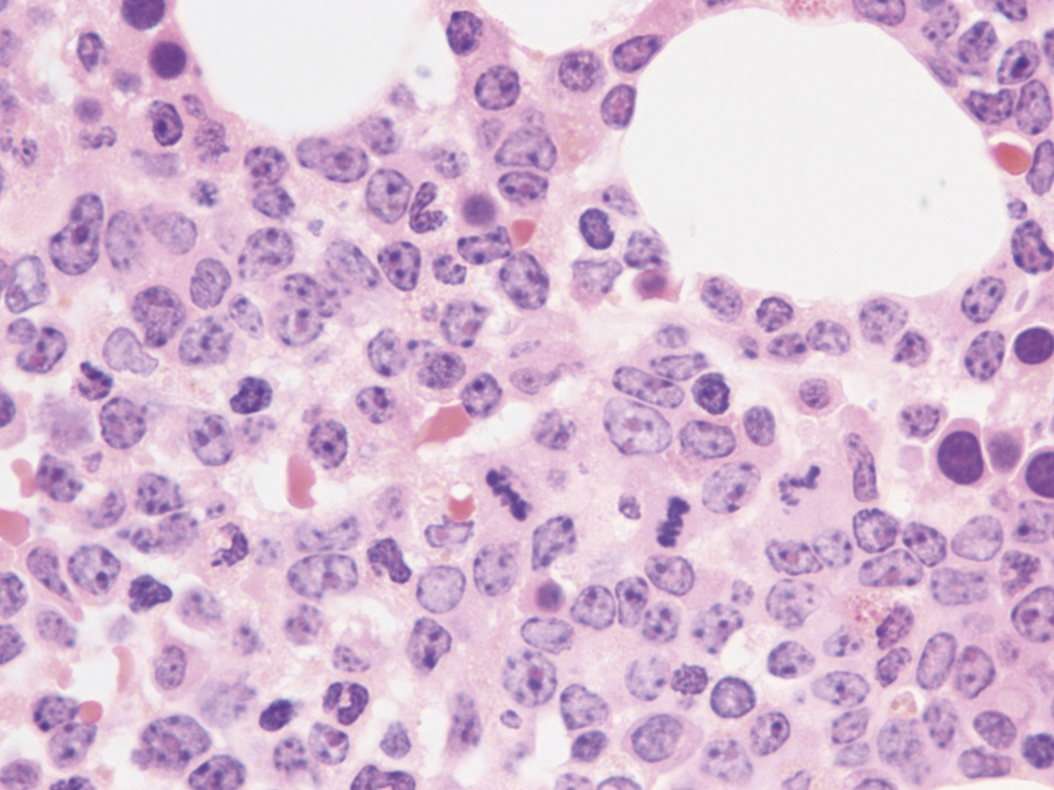
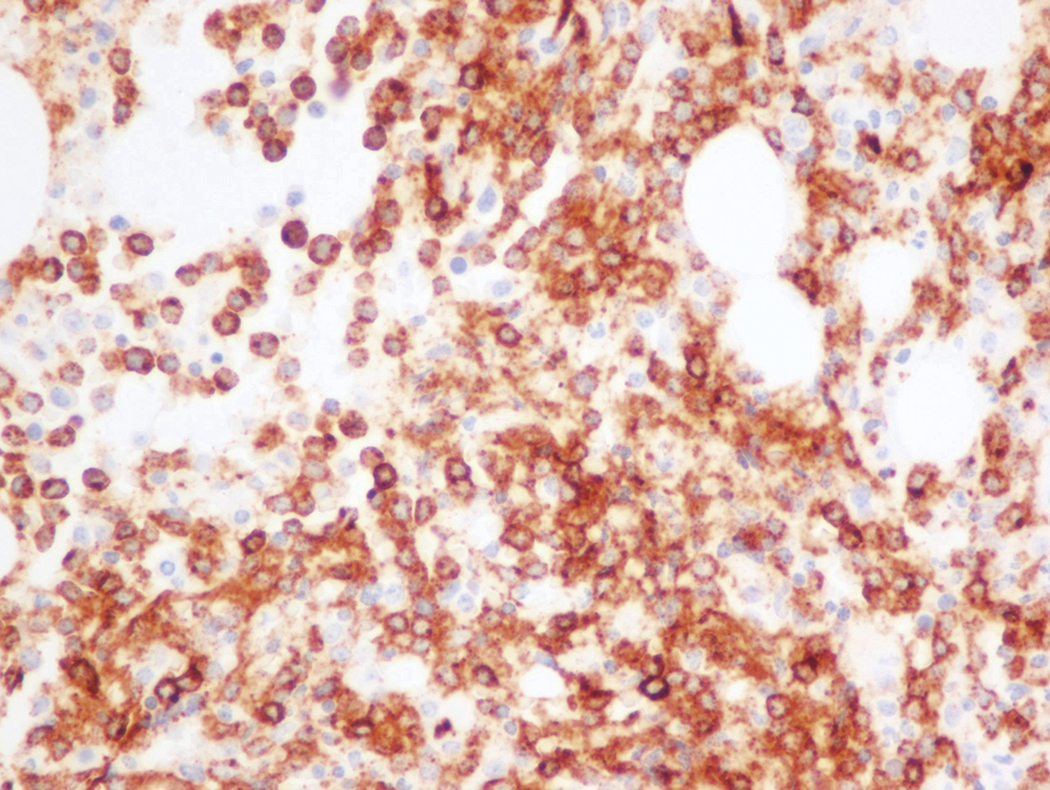
Review of the peripheral blood smear typically demonstrates thrombocytopenia of varying severity as well as a normocytic normochromic anemia ( Table 8.3 ). Normal mature granulocytes are often reduced in number. As noted earlier, a proliferation of monocytes may be seen in monocytic leukemia (AML M5), and eosinophilia may be suggestive of particular cytogenetic findings such as inv(16) or PDGFR rearrangement. , Circulating myeloid blasts most often are visible, although they may vary in both number and appearance, and may be difficult to distinguish from lymphoid blasts or atypical mature granulocytes in some instances. No single morphologic characteristic definitively identifies a myeloid blast, although certain properties are common and recurrent ( Figs. 8.7–8.17 ). Myeloid blasts may be distinguished from lymphoid blasts based on their more prominent nuclei, more open (disaggregated) chromatin structure, and the presence of Auer rods or cytoplasmic granules (although these may be relatively rare findings). In the past, myeloperoxidase staining was used to help differentiate myeloid and lymphoid blasts; however, this assay has fallen out of favor with the advent of more accurate and reliable methods such as flow cytometry (FC).
| Hematopathologic Features | |
|---|---|
| Myeloid blasts |
|
| Flow cytometry |
|
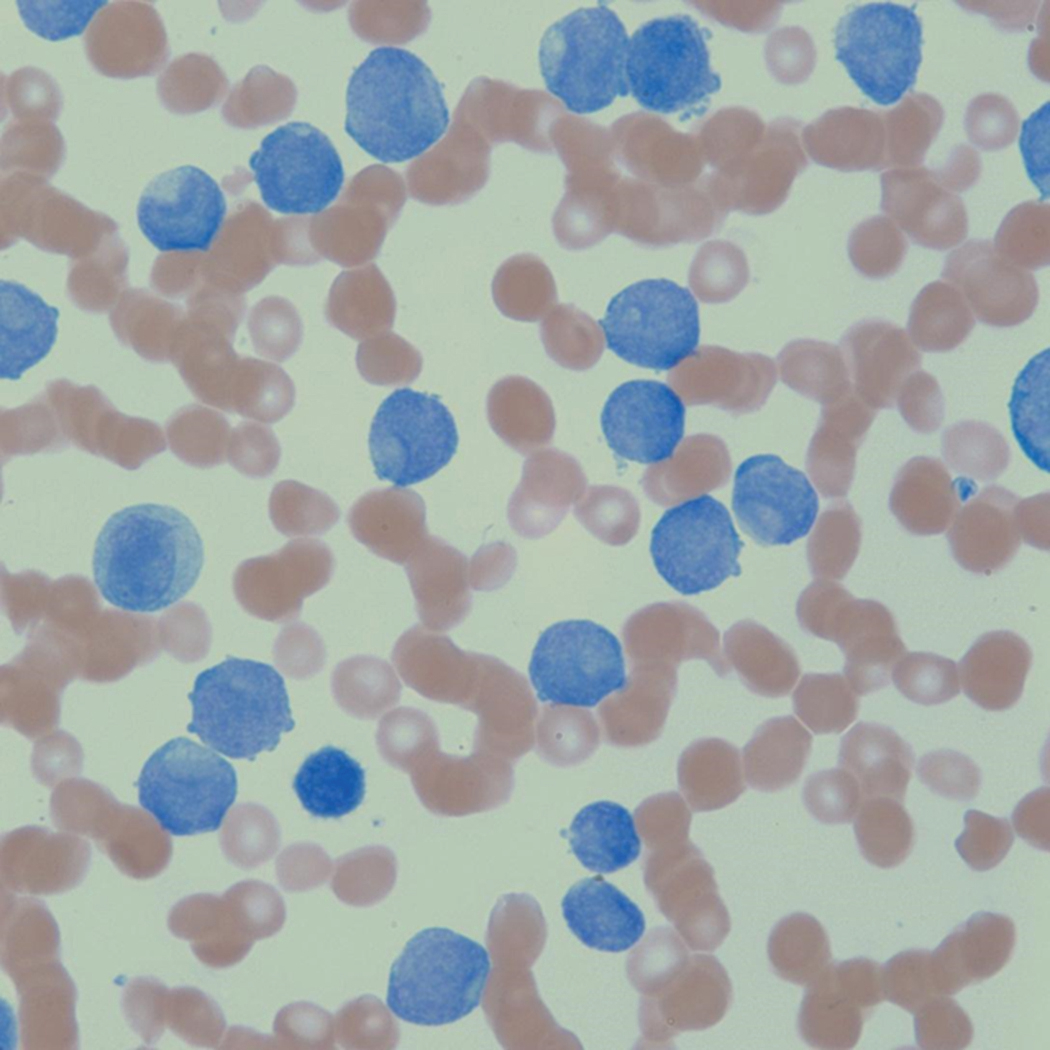
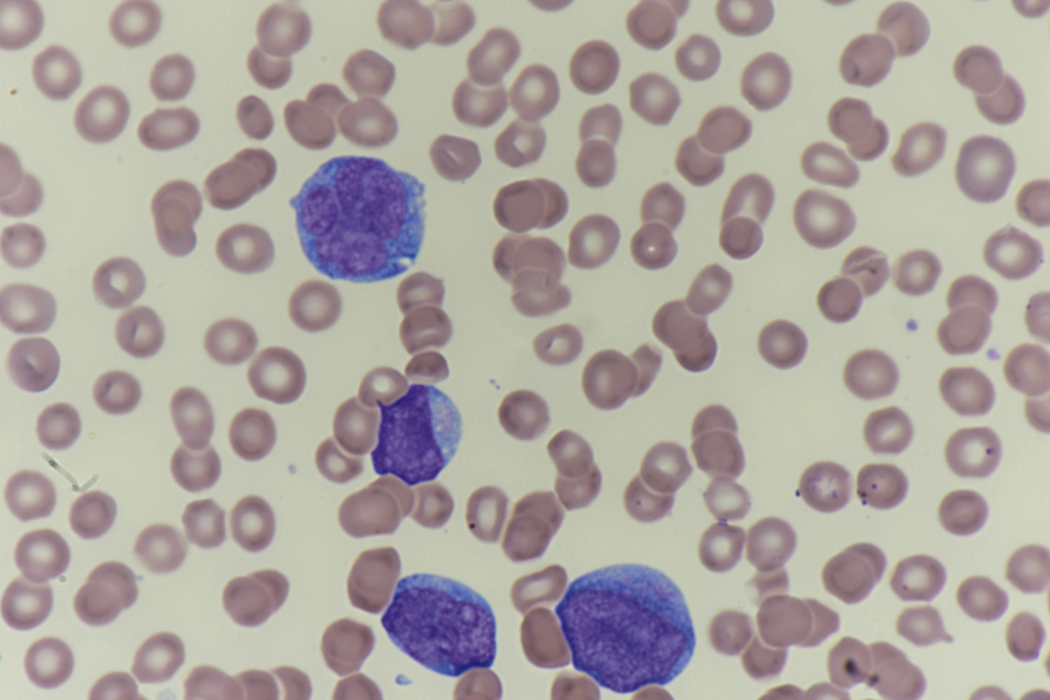

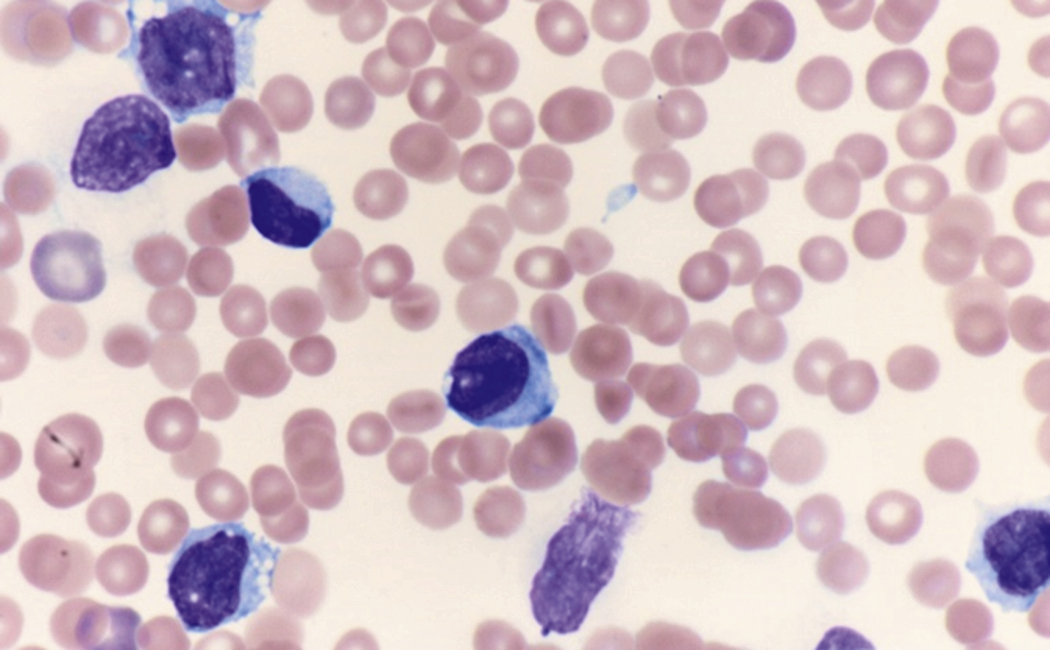
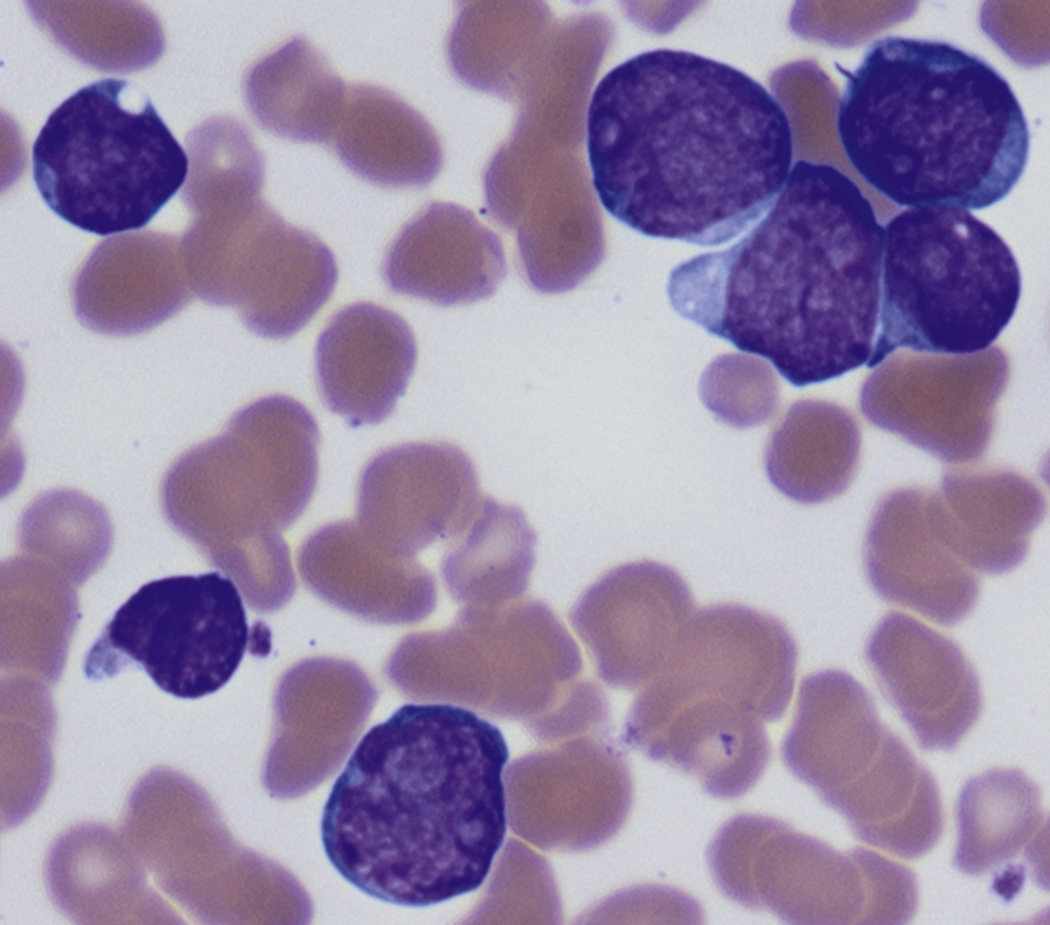
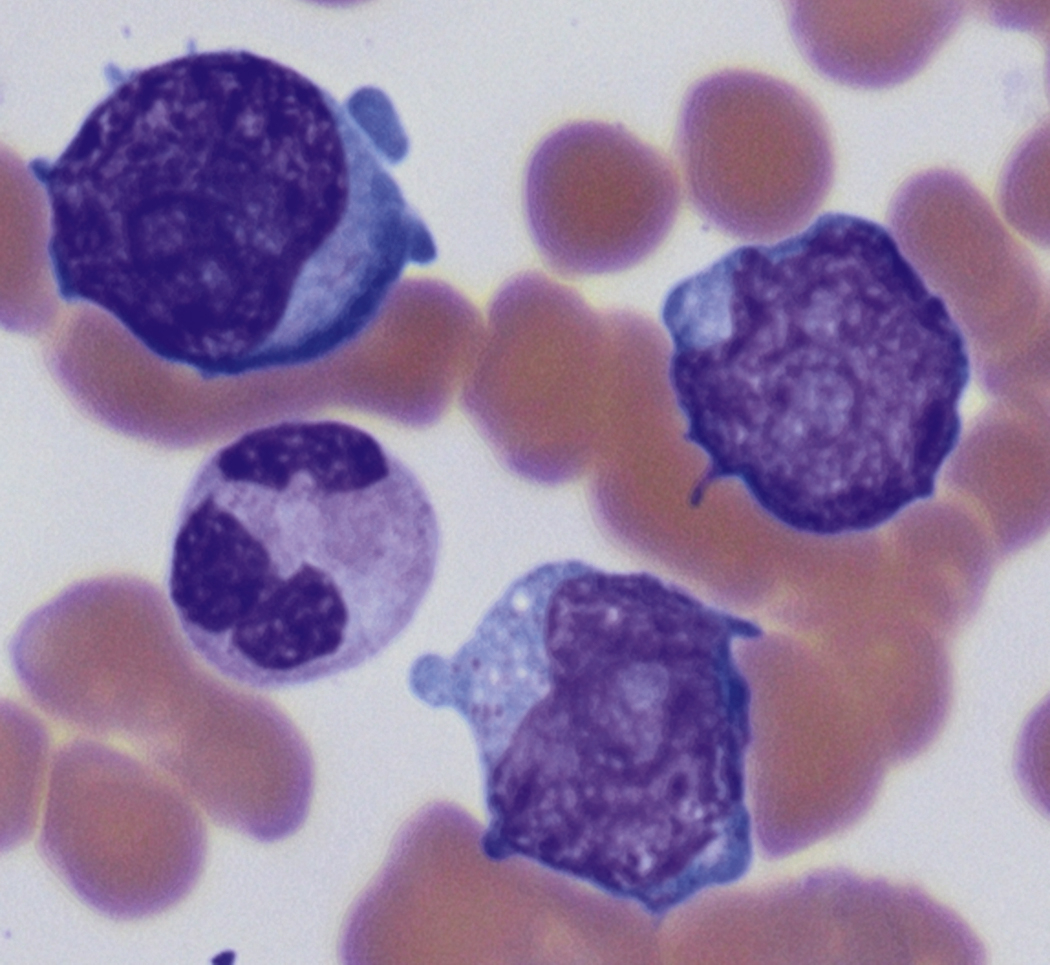
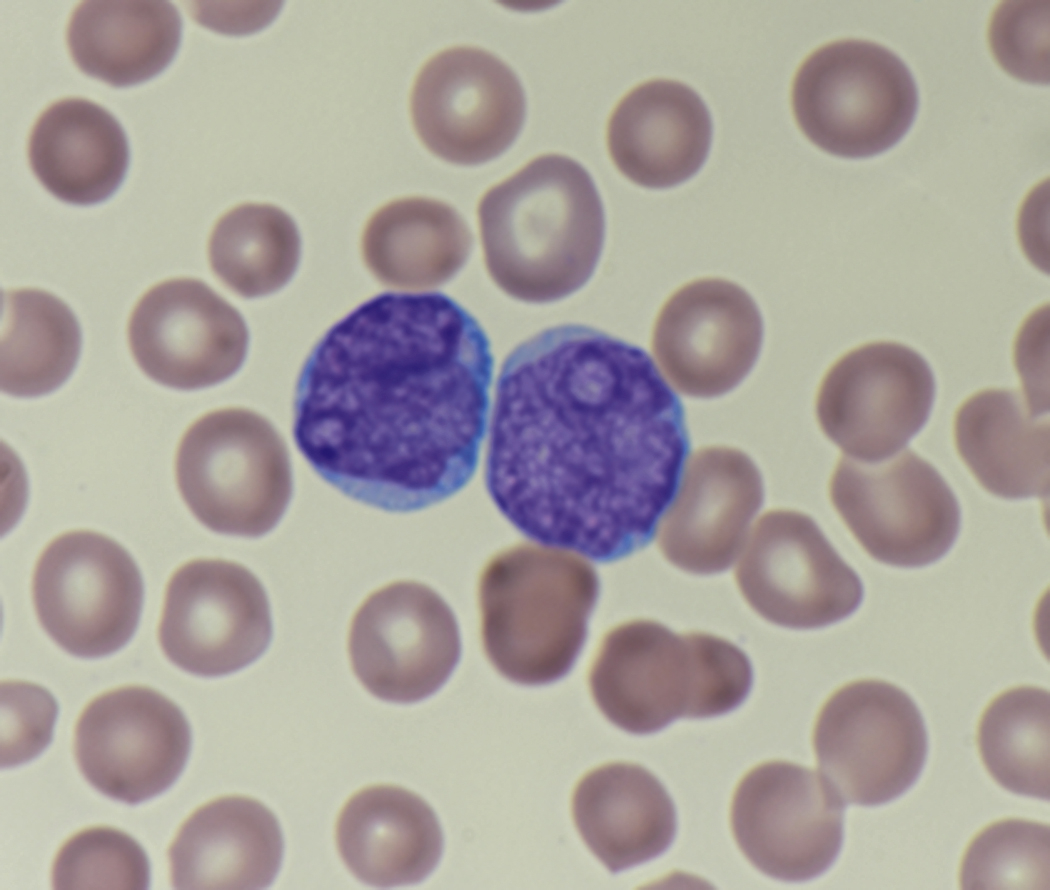
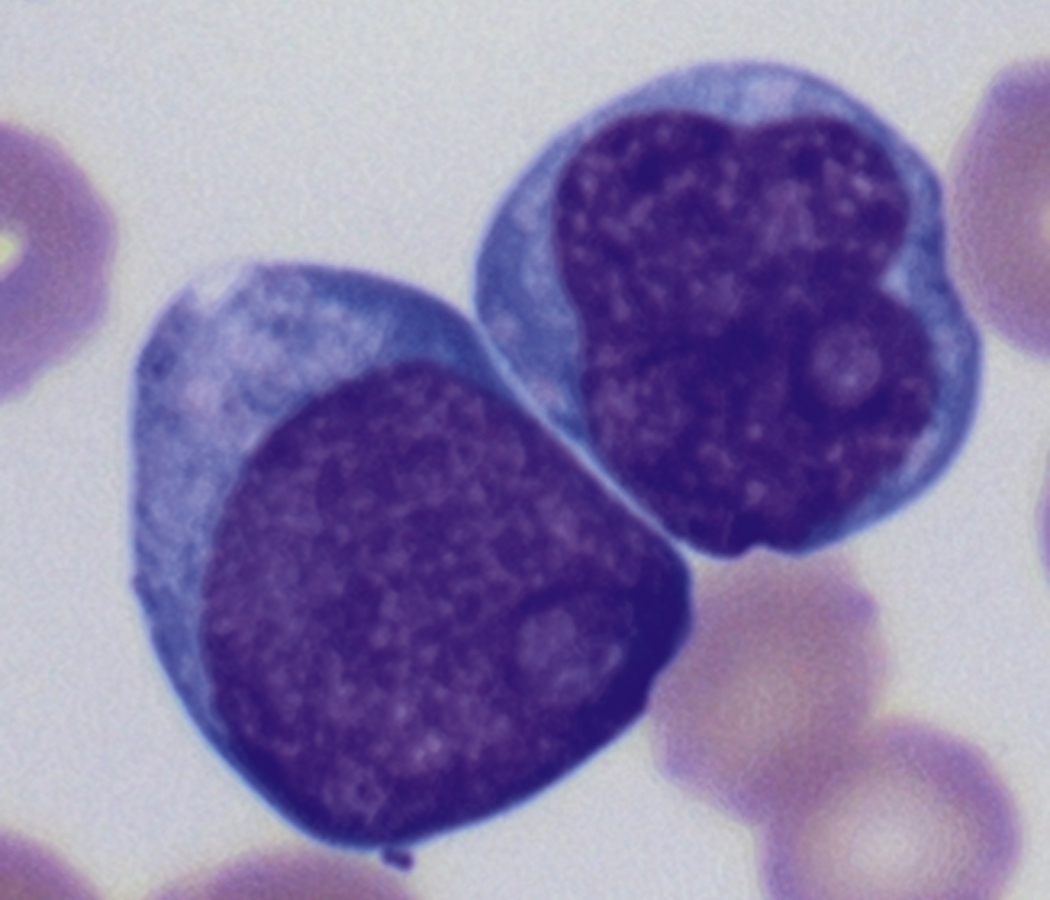
Flow cytometric analysis of peripheral blood (or bone marrow aspirate) provides a rigorous and definitive means of establishing the lineage and degree of maturation of suspected blasts, thus eliminating much of the uncertainty implicit in purely morphologic assessments ( Fig. 8.18 ).
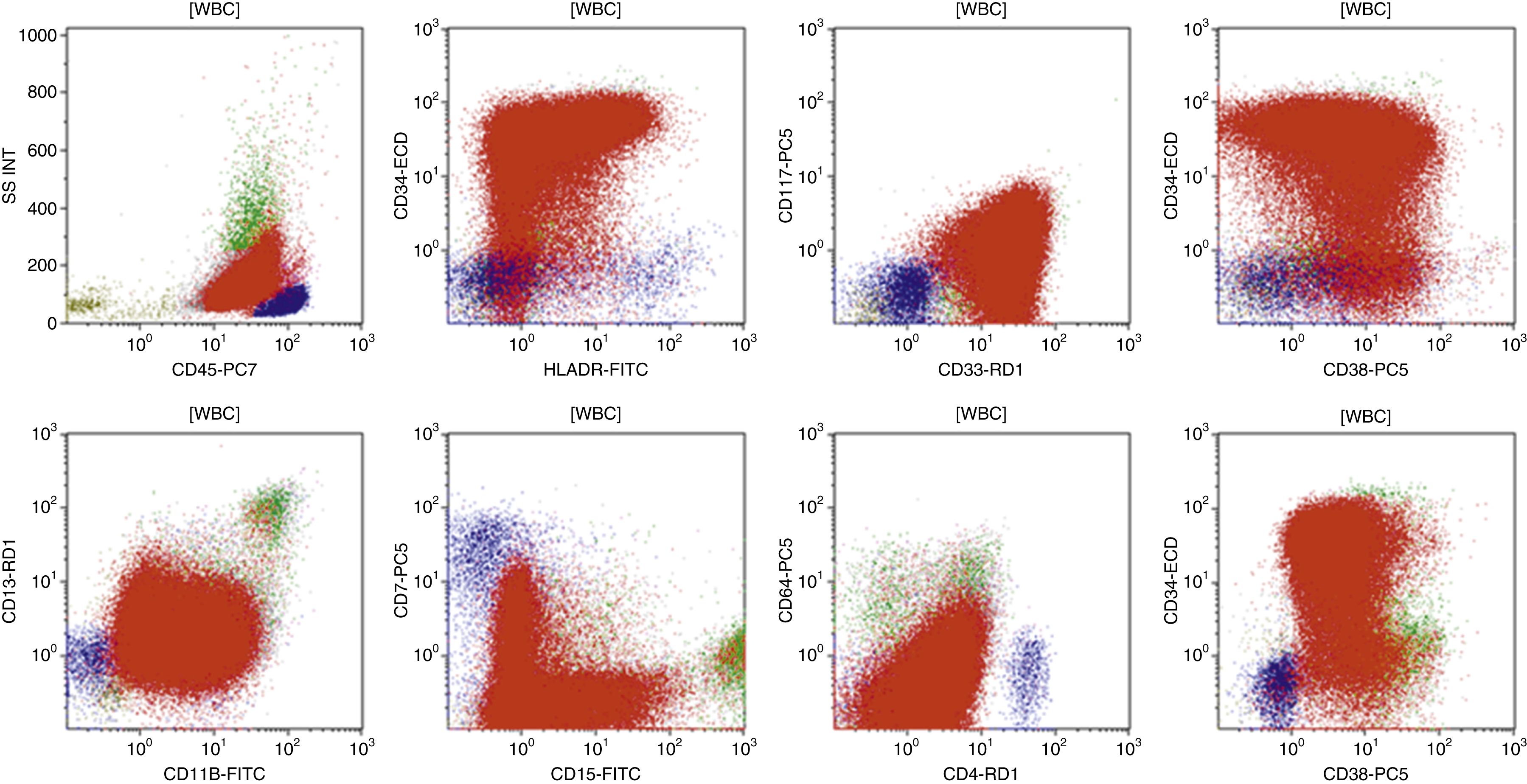
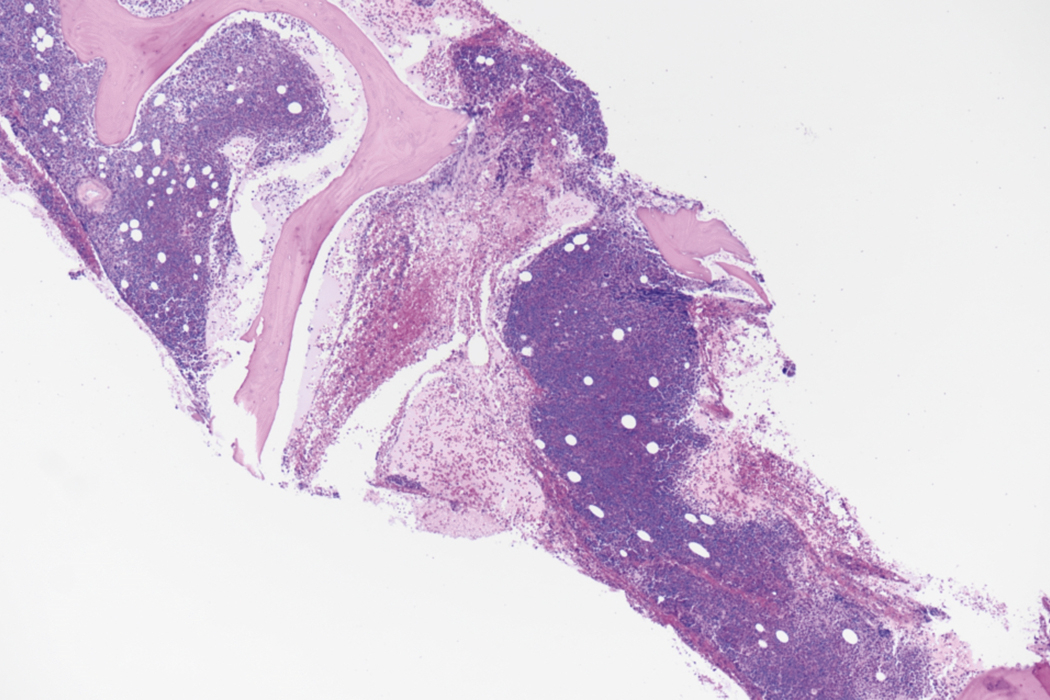
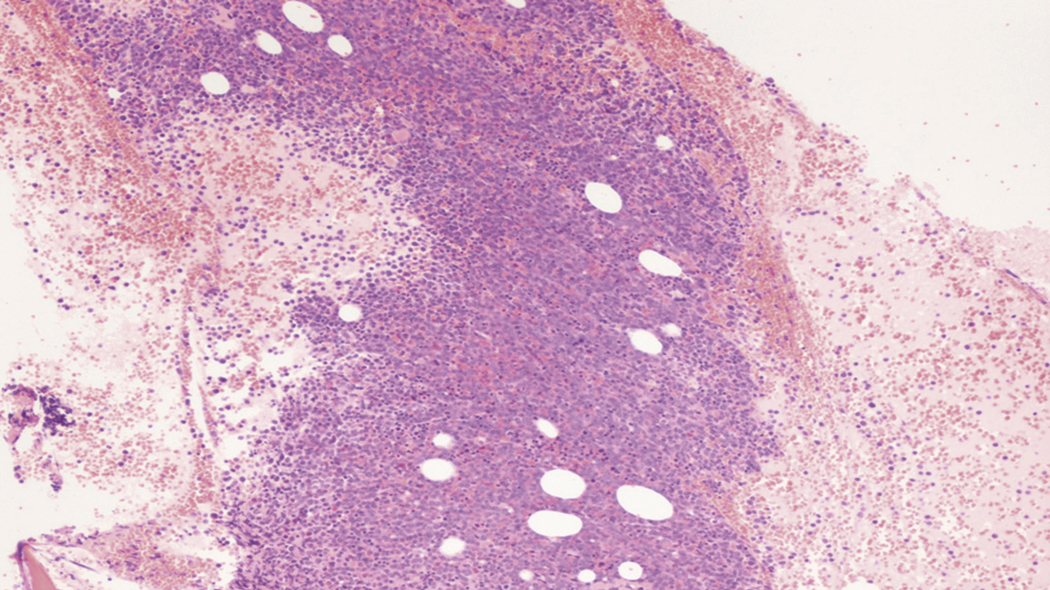
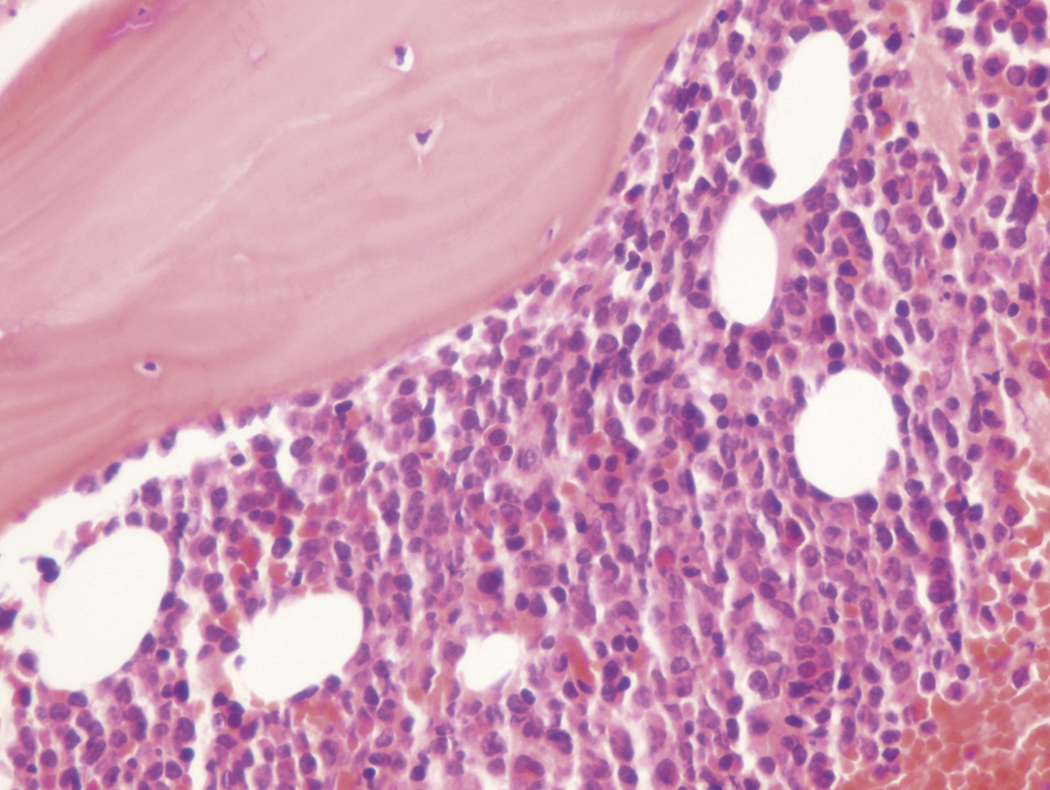
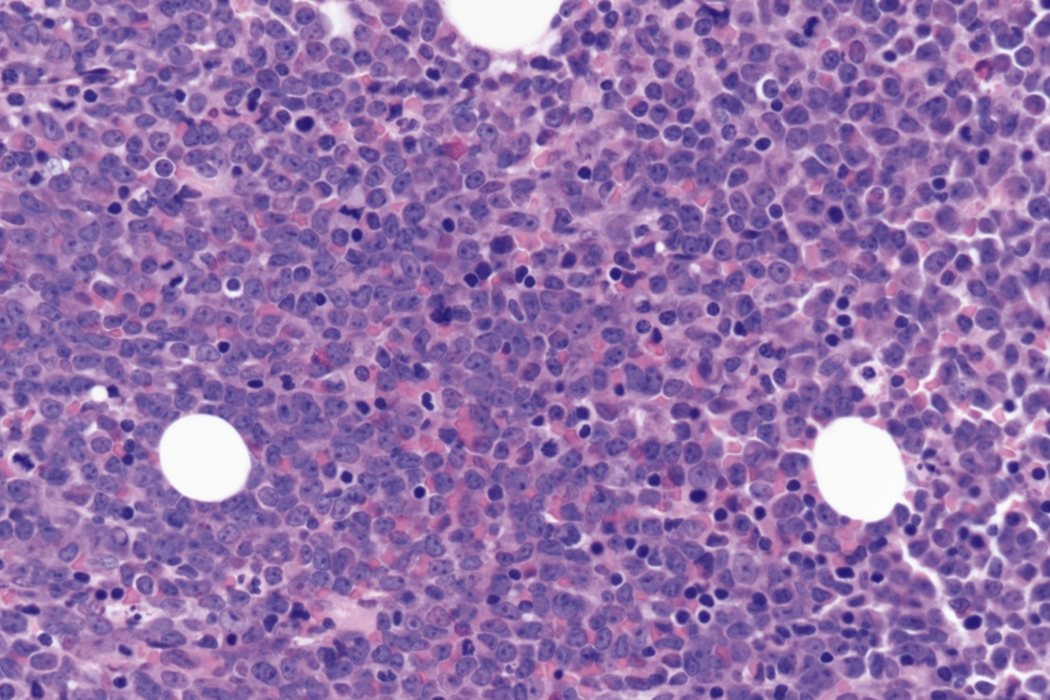
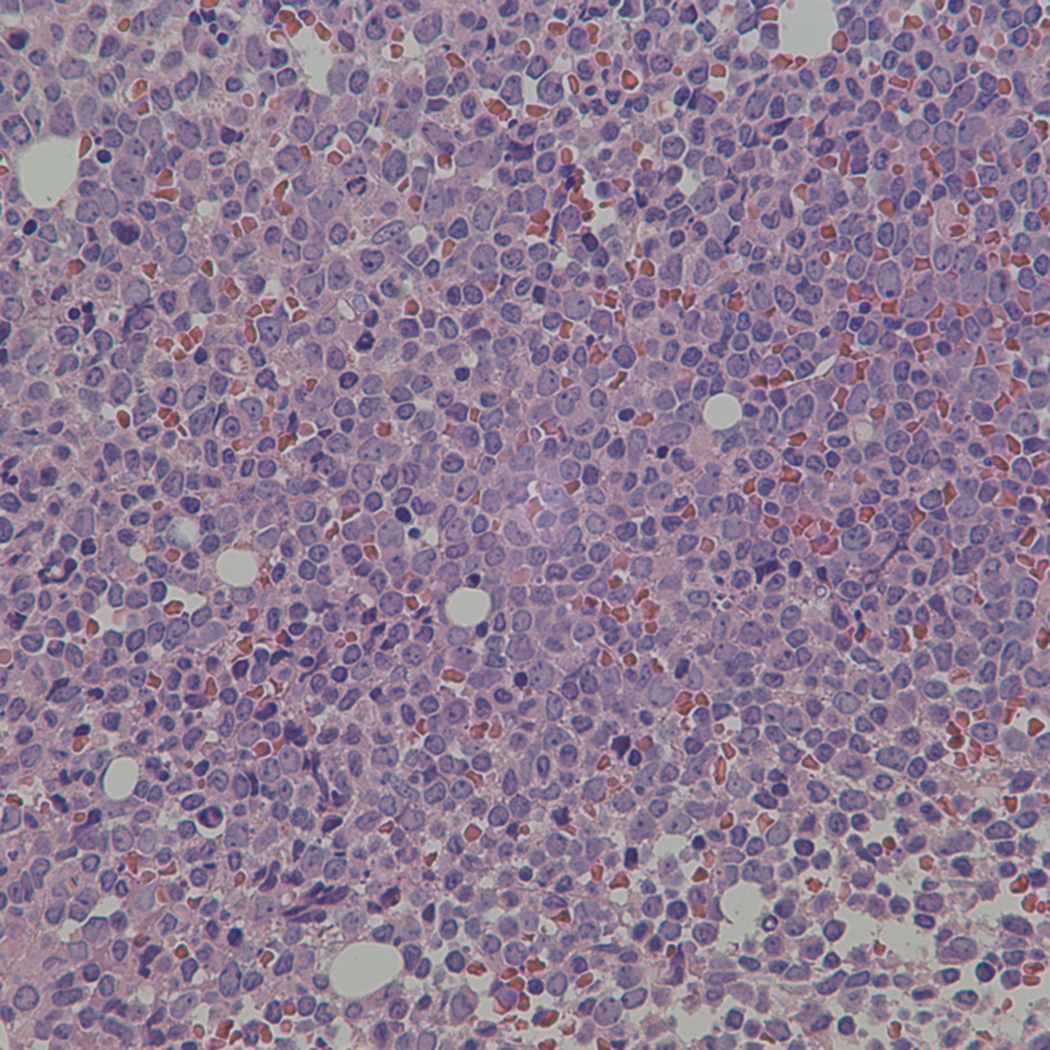
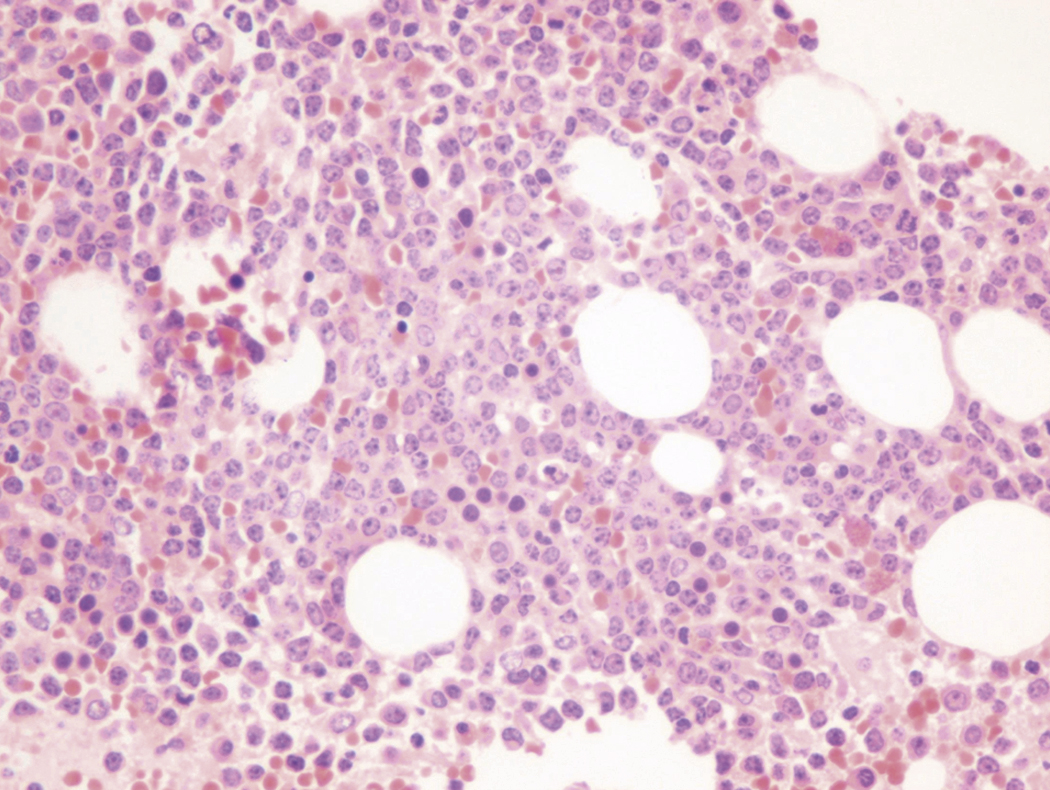
Bone marrow biopsy and aspirate are essential for the evaluation of suspected AML as a definitive diagnosis requires that blast forms account for at least 20% of bone marrow cellularity (with the notable exceptions of cases involving myeloid sarcoma or demonstrating the specific cytogenetic features t(8;21), inv(16), or t(15;17), which are deemed AML regardless of marrow blast percentage). The marrow is most often hypercellular owing to the aggressive proliferation of leukemic blasts (although instances with hypocellular marrow do occur, particularly in therapy-related cases). Much of the hematopoietic parenchyma is replaced with monotonous sheets of blasts ( Figs. 8.19–8.31 ). Blasts ( Figs. 8.32–8.37 ) may include myeloblasts, monoblasts, promonocytes, abnormal promyelocytes, and megakaryoblasts. Pronormoblasts are blast equivalents in pure erythroid leukemia. Prominent dysplasia (including dysgranulopoiesis, dyserythropoiesis, and/or dysmegakaryopoiesis) may be indicative of a preexisting MDS progressed to AML with myelodysplastic-related changes (MRCs) ( Figs. 8.38 and 8.39 ). Proliferation of stromal elements may also be present, most notably fibrosis, which may indicate a preexisting MPN (such as myelofibrosis). Of note, prominent fibrosis may also be a feature of eosinophilic leukemias (see Figs. 8.28–8.30 ) .
Case 5
A 65-year-old woman presents with fatigue, fevers, and petechiae. She is found to be pancytopenic. Manual review of her blood smear demonstrates multiple large immature-appearing cells, with immense nuclei containing lacy disaggregated chromatin, and scant basophilic agranular cytoplasm (see Fig. 8.10 ). Peripheral blood FC demonstrates an aberrant cell population that is positive for myeloid blast markers including CD34, CD33, HLADR ,CD117, and CD13, and negative for lymphoid blast markers including CD19, CD20, CD22, and tonoplast dicarboxylate transporter (TdT). A provisional diagnosis of AML is made and the results of a bone marrow aspirate and biopsy are awaited to confirm this very likely diagnosis.
Case 6
A 34-year-old man is referred to the emergency department by his primary care physician for new leukocytosis, anemia, and thrombocytopenia. Review of systems is notable for a recent decrease in exercise tolerance but is otherwise unremarkable. A CBC is repeated and indeed confirms leukocytosis in excess of 50 × 10 9 /L; however, it is also notable for eosinophilia to 30%. Manual review of the blood smear confirms abundant eosinophils; however, it is also notable for the presence of large cells, with a high nuclear-to-cytoplasmic ratio, open chromatin with multiple prominent nucleoli, and fine basophilic cytoplasm with visible nucleoli (see Fig. 8.16 ). AML with eosinophilia is suspected and the possibilities of underlying inv(16) or PDGFRA translocation are considered (as both of these entities can present with prominent eosinophilia). A bone marrow aspirate and biopsy are obtained. The biopsy is somewhat hypercellular and demonstrates significant eosinophilia. Eosinophil precursors are plentiful and demonstrate prominent basophilic-staining granules. Myeloid blast forms are present but account for less than 20% of nucleated cells. Cytogenetic studies results are eventually available and demonstrate the presence of inv(16), thus definitively establishing a diagnosis of AML. The patient’s young age and the presence of inv(16) confer a relatively favorable prognosis (see the “Prognosis” section later).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


