INTRODUCTION
SUMMARY
Acute lymphoblastic leukemia (ALL) is a malignant disorder that originates in a single B- or T-lymphocyte progenitor. Proliferation and accumulation of clonal blast cells in the marrow result in suppression of hematopoiesis and, thereafter, anemia, thrombocytopenia, and neutropenia. Lymphoblasts can accumulate in various extramedullary sites, especially the meninges, gonads, thymus, liver, spleen, and lymph nodes. The disease is most common in children but can be seen in individuals of any age. ALL has many subtypes and can be classified by immunologic, cytogenetic, and molecular genetic methods. These methods can identify clinically important, biologic subtypes, requiring treatment approaches that differ in their use of specific drugs or drug combinations, dosages of drug, or duration of treatment required to achieve optimal results. For example, cases of childhood ALL having a hyperdiploid karyotype respond well to extended treatment with methotrexate and mercaptopurine, whereas adults whose leukemic cells contain the Philadelphia chromosome and BCR-ABL1 fusion benefit from intensive treatment that includes a tyrosine kinase inhibitor and transplantation of allogeneic hematopoietic stem cells. The relative lack of therapeutic success in adult ALL is partly related to a high frequency of cases having unfavorable genetic abnormalities and partly related to poor tolerance for intensive treatment. Nearly 90 percent of children and 40 percent of adults can expect long-term, leukemia-free survival—and probable cure—with contemporary treatment. Novel immunotherapeutic approaches are under development. Currently, emphasis is placed not only on improving the cure rate but also on improving quality of life by preventing acute and late treatment-related complications, such as second malignancies, cardiotoxicity, and endocrinopathy.
Acronyms and Abbreviations
ALL, acute lymphoblastic leukemia; ARID5B, AT-rich interactive domain 5b; ATM, ataxia-telangiectasia mutated gene; CD, cluster of differentiation; CNAs, copy number abnormalities; CSF, cerebrospinal fluid; EFS, event-free survival; FISH, fluorescence in situ hybridization; HLA, human leukocyte antigen; MRI, magnetic resonance imaging; PCR, polymerase chain reaction; RT-PCR, reverse transcriptase polymerase chain reaction; SEER, Surveillance, Epidemiology, and End Results; SNP, single nucleotide polymorphism.
DEFINITION AND HISTORY
Acute lymphoblastic leukemia (ALL) is a neoplastic disease that results from multistep somatic mutations in a single lymphoid progenitor cell at one of several discrete stages of development. The immunophenotype of leukemic cells at diagnosis reflects the level of differentiation achieved by the dominant clone. The clonal origin of ALL has been established by cytogenetic analysis, by analysis of restriction fragments in female patients who are heterozygous for polymorphic X chromosome-linked genes, and by analysis of rearrangements of T-cell receptor or immunoglobulin genes. Leukemic cells divide more slowly and require more time to synthesize DNA than do normal hematopoietic counterparts. However, leukemic cells accumulate relentlessly because of their altered response to growth and death signals.1,2 They compete successfully with normal hematopoietic cells, resulting in anemia, thrombocytopenia, and neutropenia. At diagnosis, leukemic cells not only have replaced normal marrow cells but also have disseminated to various extramedullary sites.
Velpeau3 is generally credited with the earliest report of leukemia in 1827. Virchow,4 Bennett,5 and Craigie6 recognized the condition as a distinct entity by 1845. In 1847, Virchow coined the term “weisses blut” and, later, “leucaemie,” applying it to two distinct types of the disease—splenic and lymphatic—that could be distinguished from each other based on splenomegaly and enlarged lymph nodes and on the morphologic similarities of the leukemic cells to those normally found in these organs.7 Ehrlich’s introduction of staining methods in 1891 allowed further distinction of leukemia subtypes.8 By 1913, leukemia could be classified as acute or chronic, and as lymphatic or myelogenous.9 The greater prevalence of ALL in children, especially those ages 1 to 5 years, was recognized in 1917.10
Shortly after leukemia was recognized as a discrete disease entity, physicians began using chemicals as palliative therapy. The first advance was the use of a 4-amino analogue of folic acid (aminopterin), prompted by Farber’s observation that folic acid appeared to accelerate the proliferation of leukemic cells. Strikingly, for the first time, complete clinical and hematologic remissions that lasted for several months were seen in children.11 A year after the report of aminopterin-induced clinical remissions, a newly isolated adrenocorticotrophic hormone was reported to induce prompt, though brief, remissions in patients with leukemia.12 Almost concurrently, Elion and colleagues synthesized antimetabolites that interfere with synthesis of purines and pyrimidines.13 Their findings led to the introduction of mercaptopurine, 6-thioguanine, and allopurinol into clinical use. From 1950 to 1960, many new antileukemic agents were introduced, and occasional cures were seen. Pinkel and colleagues at St. Jude Children’s Research Hospital, in 1962, devised a “total therapy” approach, consisting of four treatment phases: remission induction; intensification or consolidation; therapy for subclinical CNS leukemia (or preventive meningeal treatment); and prolonged continuation therapy.14 By the early 1970s, as many as 50 percent of children achieved long-term event-free survival (EFS) using this innovative strategy. During the same period, a better understanding of the genetics of human histocompatibility and wider use of human leukocyte antigen (HLA) typing culminated in the successful use of hematopoietic stem cell transplantation for treatment of patients in whom leukemia relapsed. In the early 1980s, Riehm and coworkers introduced a so-called reinduction or delayed intensification treatment during early continuation therapy, consisting mainly of repetition of the initial remission induction and early intensification phases, and further improved the EFS to approximately 70 percent.15 Parallel to advances in treatment has been the improved understanding of the biology of ALL. The recognition of ALL as a heterogeneous group of diseases—clinically, immunologically, and genetically16—set the stage for risk-directed therapy.16a
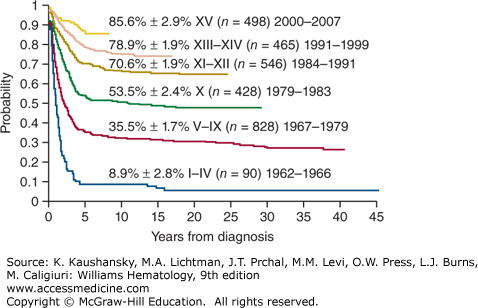
Treatment of ALL has progressed incrementally, beginning with the development of effective therapy for CNS disease, followed by intensification of early treatment, especially for patients at high risk of relapse. The current cure rates of nearly 90 percent for children (Fig. 91–1) and 40 percent for adults attest to the steady progress made in treating this disease.17,18 Rapid evolution and convergence of multiple genome-wide platforms to identify the total complement of genetic and epigenetic alterations almost certainly will lead to the identification of new targets for specific treatment.19,20 A clear advance was the development of imatinib mesylate and dasatinib, which target leukemias with the BCR-ABL1 fusion.21
Figure 91–1.
Kaplan-Meier analysis of event-free survival for 2855 children with ALL treated in 15 consecutive total-therapy studies at St. Jude Children’s Research Hospital. Early intensification of systemic and intrathecal chemotherapy with a risk assignment based on sequential measurements of minimal residual disease in the 2000s has boosted the event-free survival estimate to 85.6 percent ± 2.9 percent (SE). (Data from CH Pui and is unpublished.)
ETIOLOGY AND PATHOGENESIS
Initiation and progression of ALL are driven by successive mutations that alter cellular functions, including an enhanced ability of self-renewal, a subversion of control of normal proliferation, a block in differentiation, and an increased resistance to death signals (apoptosis).1,2 Familial disorders of DNA repair may play a role. Environmental agents, such as ionizing radiation and chemical mutagens, have been implicated in the induction of ALL in some patients. However, in most cases, no etiologic factors are discernible. In the favored theory, leukemogenesis reflects the interaction between host pharmacogenetics (susceptibility) and environmental factors, a model that requires confirmation in well-designed population and molecular epidemiologic studies.
The American Cancer Society has estimated that in the United States there will be approximately 6020 new cases of ALL in 2014 (3140 in males and 2880 in females) and approximately 1440 deaths from ALL (810 in males and 630 in females).22 Most cases of ALL occur in children, but most deaths from ALL (approximately four of five) occur in adults.
The age-adjusted incidence rate of ALL was 1.6 per 100,000 males and 1.2 for females per year in the United States, based on cases diagnosed in 1975 to 2010 from 17 Surveillance, Epidemiology, and End Results (SEER) geographic areas.23 The risk for developing ALL is highest in children younger than 5 years of age. The risk then declines slowly until the mid-20s, and begins to rise again slowly after age 50. The incidence is 7.9 per 100,000 children 1 to 4 years old and 1.2 for those older than age 60 years. Only 20 percent of adult acute leukemias are ALL, but about one-third of ALL cases are in adults. The average person’s lifetime risk of developing ALL is less than one in 750. The risk is slightly higher in males than in females, and higher in whites than in African Americans (Fig. 91–2).23 The median age at diagnosis for ALL is 13 years and approximately 61 percent are diagnosed before the age of 20 years, however, because of the bimodal peak in incidence, the age of 13 years is mathematically correct but medically nearly useless. ALL is the most common malignancy diagnosed in patients younger than age 15 years, accounting for 23 percent of all cancers and 76 percent of all leukemias in this age group.
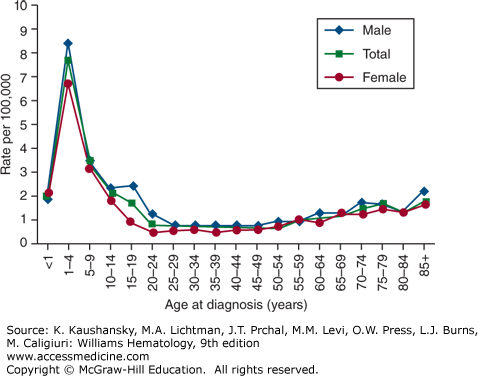
The sharp incidence peak of ALL during early childhood has been observed only since the 1930s in the United Kingdom and the United States.24 In the United States, the peak first appeared in children of European descent, and subsequently was seen in children of African descent in the 1960s. The age peak is absent in many developing or underdeveloped countries, suggesting a leukemogenic contribution from factors associated with industrialization. Except for a slight predominance for females in infancy, ALL affects males of European descent more often than females in all age groups (see Fig. 91–2). The frequency distribution is similar among those of African descent. In most age groups, the incidence of ALL is higher in those of European descent than in those of African descent, especially among children ages 2 to 3 years.
The incidence of ALL differs substantially in different geographic areas. Rates are higher among populations in northern and western Europe, North America, and Oceania, with lower rates in Asian and African populations.25 In Europe, the highest rates of ALL among males are found in Spain and the highest rates among females in Denmark. In the United States, the highest rates for both sexes are among Latinos in Los Angeles.
The precise pathogenetic events leading to the development of ALL are unknown. Only a minority (5 percent) of cases are associated with inherited, predisposing genetic syndromes. Children with Down syndrome have a 10 to 30 times greater risk of leukemia; acute megakaryoblastic leukemia predominates in those patients younger than age 3 years, and ALL is predominant in older age groups. ALL in patients with Down syndrome is a heterogeneous disorder, comprising subtypes with the same well-recognized genetic abnormalities found in the general population, such as hyperdiploidy greater than 50 and t(12;21)[ETV6-RUNX1], plus those more commonly associated with Down syndrome such as +X, del(9), and CEBPD rearrangement.26,27 Studies show that P2RY8-CRLF2 fusion and activating JAK mutations together contribute to leukemogenesis in approximately half of the cases of Down syndrome patients with ALL.28,29 Almost all ALL patients with Down syndrome have a deletion of IKZF1.30 Autosomal recessive genetic diseases associated with increased chromosomal fragility and a predisposition to ALL include ataxia-telangiectasia, Nijmegen breakage syndrome, and Bloom syndrome.31 Patients with ataxia-telangiectasia have a 70 times greater risk of leukemia and a 250 times greater risk of lymphoma, particularly of the T-cell phenotype.32 The causative gene, termed ATM (ataxia-telangiectasia mutated), encodes a protein involved in DNA repair, regulation of cell proliferation, and apoptosis. Laboratory studies supporting the diagnosis of ataxia-telangiectasia include an elevated serum concentration of α-fetoprotein, presence of characteristic chromosomal aberrations, absent or reduced intranuclear serine protein kinase ATM, and increased in vitro radiosensitivity. A high prevalence of germline truncating and missense ATM gene alterations in children with sporadic T-cell ALL suggests a pathogenetic role of ATM in lymphoid malignancies. Although impaired immune surveillance contributes to the increased risk of Epstein-Barr virus-related malignancies in patients with acquired immunodeficiencies, no compelling evidence indicates defective immunity contributes to the predisposition to ALL in patients with ataxia-telangiectasia or other congenital immunodeficiency syndromes. Genome-wide association studies have identified common allelic variants in four genes (IKZF1, ARID5B, CEBPE, and CDKN2a) that are consistently associated with childhood ALL.33,34,35 These genes are key regulators of blood cell development, and acquired mutations of each are also detected in ALL cases. Thus, the risk of childhood ALL may be influenced by coinheritance of multiple low-risk variants. Inherited allelic variation may also affect response to treatment.36
In utero (but not postnatal) exposure to diagnostic x-rays confers a slightly increased risk of ALL, which correlates positively with the number of exposures.37 The evidence is weak for an association between the development of ALL and nuclear fallout; exposure to occupational, natural terrestrial, or cosmic ionizing radiation exposure; or paternal radiation exposure prior to conception. There has been concern that exposure to low-energy electromagnetic fields produced by a residential power supply may be associated with the development of childhood ALL. Case-control studies suggested a slightly increased risk of leukemia at very high levels of exposure; assuming the association is real, only approximately 1 percent of leukemias could be attributed to the exposure.38,39 Pesticide exposure (occupational or home use) and parental cigarette smoking before or during pregnancy, administration of vitamin K to neonates, maternal alcohol consumption during pregnancy, and increased consumption of dietary nitrites have each been suggested causes. However, each of these associations is controversial, and most have been refuted after careful, controlled investigation. High birth weight is associated with an increased risk of leukemia before the age of 5 years with fair consistency,40 and the birth weight is likely a marker for an endogenous factor, such as insulin-like growth factor.
Subtle genetic polymorphisms of xenobiotic-metabolizing enzymes, DNA repair pathways, and cell-cycle checkpoint functions might interact with environmental, dietary, maternal, and other external factors to affect the development of ALL.2,41 Although the number of investigations and sample sizes are limited, data exist to support a causal role for polymorphisms in genes encoding detoxifying enzymes (e.g., glutathione S-transferase, nicotinamide adenine dinucleotide phosphate [NAD(P)H]:quinone oxidoreductase), folate-metabolizing enzymes (serine hydroxymethyltransferase and thymidylate synthase), cytochrome P450, methylenetetrahydrofolate reductase, and cell-cycle inhibitors in the development of adult and childhood ALL.42,43 However, all these associations must be confirmed by larger studies with careful attention to ethnic and geographic diversity in the frequency of polymorphisms. Using genome-wide analysis, germline single-nucleotide polymorphisms (SNPs) of AT-rich interactive domain 5b (ARID5B) gene have been associated with childhood hyperdiploid B-cell precursor ALL,44 a clear example of host genetic variations affecting the susceptibility to the development of childhood ALL.
Retrospective identification of leukemia-specific fusion genes (e.g., KMT2A/AFF1 [also known as MLL-AF4] and ETV6-RUNX1 [also known as TEL-AML1]), hyperdiploidy, or clonotypic rearrangements of immunoglobulin or T-cell receptor loci in archived neonatal blood spots (Guthrie cards), and development of concordant leukemia in identical twins clearly indicate some leukemias have a prenatal origin.45,46 In identical twins with the t(4;11)/KMT2A/AFF1, the concordance rate is nearly 100 percent, and the latency in the time of occurrence in the two twins is short (a few weeks to a few months). These findings suggest this fusion gene alone either is leukemogenic or requires only a small number of cooperative mutations to cause leukemia. By contrast, the lower concordance rate in twins with the ETV6-RUNX1 fusion or T-cell phenotype and the longer postnatal latency period suggest additional postnatal events are required for leukemic transformation in these subtypes.45 This theory is supported by the identification of rare cells expressing ETV6-RUNX1 fusion transcripts in approximately 1 percent of cord blood samples from newborns, a frequency 100 times higher than the incidence of ALL defined by this fusion transcript.45 The presence of a preleukemic clone with the ETV6-RUNX1 has been established.47 Hyperdiploid ALL, another common subtype of childhood ALL, also appears to arise before birth but requires postnatal events for full malignant transformation.46 The observations of a peak age of development of childhood ALL of 2 to 5 years, an association of industrialization and modern or affluent societies with increased prevalence of ALL, and the occasional clustering of childhood leukemia cases have fueled two parallel infection-based hypotheses to account for postnatal events. The “delayed-infection” hypothesis suggests that some susceptible individuals with a prenatally acquired preleukemic clone had low or no exposure to common infections early in life because they lived in an affluent hygienic environments.45 Such infectious insulation predisposes the immune system of these individuals to aberrant or pathologic responses after subsequent or delayed exposure to common infections at an age commensurate with increased lymphoid cell proliferation. The “population-mixing” hypothesis predicts that clusters of childhood ALL result from exposure of susceptible (nonimmune) individuals to common but fairly nonpathologic infections after population mixing with carriers.48 However, clearly not all childhood cases develop in utero. For example, t(1;19)/TCF3-PBX1 (also known as E2A-PBX1) ALL appears to have a postnatal origin in most cases.49 Cases of adult ALL most certainly arise over a protracted time.
Acquired genetic abnormalities are a hallmark of ALL; 80 percent of all cases have recurring cytogenetic or molecular lesions with prognostic and therapeutic relevance (Table 91–1).2,19,41 Chromosomal changes include abnormalities in the number (ploidy) and structure of chromosomes.50,51,52 The latter comprise translocations (the most frequent abnormality), inversions, deletions, point mutations, and amplifications. Although the frequency of particular genetic subtypes differs between childhood and adult cases, the general mechanisms underlying the induction are similar. Mechanisms include aberrant expression of oncoproteins, loss of tumor-suppressor genes, and chromosomal translocations that generate fusion genes encoding transcription factors or active kinases.
| Abnormality | Children (%) | Adults (%) |
|---|---|---|
| Hyperdiploidy (>50 chromosomes) | 23–29 | 6–7 |
| Hypodiploidy (<45 chromosomes) | 1 | 2 |
| t(1;19)(q23;p13.3) [TCF3-PBX1] | 4 in white, 12 in black | 2–3 |
| t(9;22)(q34;q11.2) [BCR-ABL1] | 2–3 | 25–30 |
| t(4;11)(q21;q23) [MLL-AF4] | 2 | 3–7 |
| t(8;14)(q23;q32.3) | 2 | 4 |
| t(12;21)(p13;q22) [ETV6-RUNX1] | 20–25 | 0–3 |
| NOTCH1 mutations* | 7 | 15 |
| HOX11L2 overexpression* | 20 | 13 |
| LYL1 overexpression* | 9 | 15 |
| TAL1 overexpression* | 15 | 3 |
| HOX11 overexpression* | 7 | 30 |
| MLL-ENL fusion | 2 | 3 |
| Abnormal 9p | 7–11 | 6–30 |
| Abnormal 12p | 7–9 | 4–6 |
| del(7p)/del(7q)/monosomy 7 | 4 | 6–11 |
| +8 | 2 | 10–12 |
| Intrachromosomal amplification of chromosome 21 (iAMP21) | 2 | ? |
Primary genetic rearrangement by itself is insufficient to induce overt leukemia. Cooperative mutations are necessary for leukemic transformation and include genetic and epigenetic changes in key growth regulatory pathways.19,20 The candidate gene approach has identified deletion of the CDKN2A/CDKN2B tumor-suppressor locus53 and mutations of NOTCH1 in T-cell ALL.54 Current searches applying genome-wide microarray and high-throughput sequencing methodologies have identified a high frequency of common genetic alterations in both B-cell precursor ALL and T-cell ALL. Using SNP microarray, a mean of 6.46 DNA copy number abnormalities (CNAs) per case was identified, suggesting that gross genomic instability is not a feature for most ALL cases.55 There was a wide variation in the number of CNAs across leukemic subtypes. Interestingly, infant ALL cases with MLL rearrangement had less than one CNA per case, suggesting that few additional genetic lesions are required for leukemogenesis in these cases. By contrast, ETV6-RUNX1 and BCR-ABL1 cases had more than six CNAs per case, with some having more than 20 lesions, a finding consistent with the concept that, although the initiating events may occur early in childhood, additional lesions are required for subsequent development of ALL. More than 40 percent of B-cell precursor ALL cases had mutations in genes encoding regulators of normal lymphoid development.55 The most frequent target was the lymphoid transcription factor PAX5 (mutated in approximately 32 percent of cases), which encode a paired-domain protein required for the pro–B-cell to pre–B-cell transition and B-lineage fidelity. The second most frequently involved gene was IKZF1 (mutated in almost 28 percent of the cases), encoding the IKAROS zinc finger DNA-binding protein that is required for the earliest lymphoid differentiation. IKZF1 was deleted in the vast majority of cases of BCR-ABL1 ALL cases as well as chronic myeloid leukemia in lymphoid blast crisis (but not chronic phase).56 Approximately half of BCR-ABL1 ALL cases also had deletions of CDKN2A/B and PAX5. This finding further supports the concept that multiple signaling pathways need to be disrupted to induce leukemia. A subgroup of ALL with very poor outcome was strongly associated with the presence of IKZF1 deletions.57,58 Together, these findings suggest that IKZF1 directly contributes to treatment resistance in ALL.
BCR-ABL1–like B-cell ALL lacks the BCR-ABL1 fusion or t(9;22) by cytogenetic, fluorescence in situ hybridization (FISH), or molecular analyses, but it shares the same gene-expression profile with typical BCR-ABL1–positive ALL.59 In half of these cases, the CRLF2 gene is involved in a cryptic translocation with the IGH gene or is fused to the P2RY8 gene; both rearrangements lead to overexpression of CRLF2.29,60 Mutations in JAK2 or JAK1 are detected in 30 to 40 percent of these cases, and many of the remaining have activating mutations in cytokine receptor and kinase signaling pathways.30 Microarrays and genomic DNA sequencing identified monoallelic deletion of the PAX5 gene at chromosome band 9p13.2 in 28 percent of ALL patients with cryptic or larger deletions on 9p.55
Gene-expression profiling with DNA microarrays allows nearly all T-cell cases to be grouped according to multistep oncogenic pathways.61 Gene-expression studies also show that overexpression of FLT3, a receptor tyrosine kinase important for development of hematopoietic stem cells, is a secondary event in almost all cases with either MLL rearrangements or hyperdiploidy.62 The finding has provided an impetus for clinical testing of FLT3 inhibitors in ALL. Other genome-wide interrogations of both leukemia cells and germline tissues have identified other genetic variations with prognostic or therapeutic relevance and may lead to the development of specific treatment.17,36
Epigenetic changes, including hypermethylation and silencing of tumor-suppressor genes and hypomethylation of oncogenes and abnormalities in posttranscriptional control mechanisms, such as those involving microRNA, are common findings in cancer. These changes are reversible and do not alter the DNA sequence, yet they can alter gene expression in subtle ways that encourage malignant transformation and progression. The analysis of epigenetic alterations has begun to apply to the development of new biomarkers for risk assignment or disease monitoring, and to the design of alternative treatment in ALL.63 Evidence indicates that the methylation of multiple genes in ALL is associated with a worse outcome. Surprisingly, methylation of genes was as prominent in childhood as in adult ALL. The differences in the response of children and adults appear not to be related to quantitative methylation but to the specific genes and the specific pathways deactivated. Preliminary studies of hypomethylating agents (e.g., azacitidine and decitabine) are being tested in patients refractory or resistant to current drug programs.64
CLINICAL FEATURES
The clinical presentation of ALL is highly variable. Symptoms may appear insidiously or acutely. The presenting features generally reflect the degree of marrow failure and the extent of extramedullary spread (Table 91–2).18,65,66,67 Approximately half of patients present with fever, which can be caused by either neutropenia-induced infection or leukemia-released cytokines (e.g., interleukin-1, interleukin-6, and tumor necrosis factor) released from leukemia cells. In these patients, fever resolves within 72 hours after the start of antileukemia therapy.
| Feature | Children (%) | Adult (%) |
|---|---|---|
| Age (years) | ||
| <1 | 2 | — |
| 1–9 | 72–78 | — |
| 10–19 | 20–26 | — |
| 20–39 | — | 40 |
| 40–59 | — | 40 |
| ≥60 | — | 20 |
| Male | 56–57 | 62 |
| Symptoms | ||
| Fever | 57 | 33–56 |
| Fatigue | 50 | Common |
| Bleeding | 43 | 33 |
| Bone or joint pain | 25 | 25 |
| Lymphadenopathy | ||
| None | 30 | 51 |
| Marked (>3 cm) | 15 | 11 |
| Hepatomegaly | ||
| None | 34 | 65 |
| Marked (below umbilicus) | 17 | Rare |
| Splenomegaly | ||
| None | 41 | 56 |
| Marked (below umbilicus) | 17 | Uncommon |
| Mediastinal mass | 8–10 | 15 |
| CNS leukemia | 3 | 8 |
| Testicular leukemia | 1 | 0.3 |
Fatigue and lethargy are common manifestations of anemia in patients with ALL. In older patients, anemia-related dyspnea and lightheadedness may be the dominant presenting features. More than 25 percent of patients, especially young children, may have a limp from bone pain or arthralgia, or an unwillingness to walk because of leukemic infiltration of the periosteum, bone, or joint or because of expansion of the marrow cavity by leukemia cells. Children with prominent bone pain often have nearly normal blood counts, which can contribute to delayed diagnosis. In a small proportion of patients, marrow necrosis can result in severe bone pain and tenderness, fever, and a very high level of serum lactate dehydrogenase.68,69 Arthralgia and bone pain are less severe in adults. Less common signs and symptoms include headache, vomiting, altered mental function, oliguria, and anuria. Occasionally, patients present with a life-threatening infection or bleeding (e.g., intracranial hematoma). Intracranial hemorrhage occurs mainly in patients with an initial leukocyte count greater than 400 × 109/L.70 Very rarely, ALL produces no signs or symptoms and is detected during routine examination.
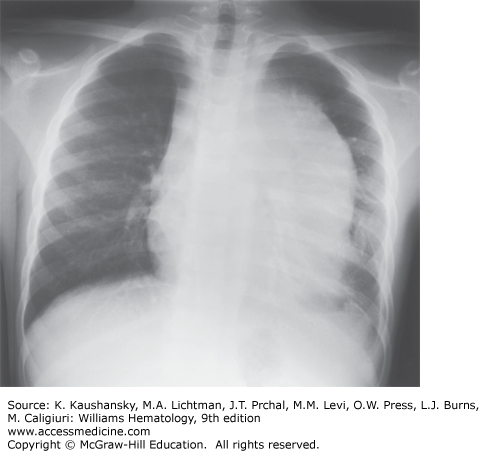
Among frequent findings are pallor, petechiae, and ecchymosis in the skin and mucous membranes, and bone tenderness as a result of leukemic infiltration or hemorrhage that stretches the periosteum. Liver, spleen, and lymph nodes are the most common sites of extramedullary involvement, and the degree of organomegaly is more pronounced in children than in adults. An anterior mediastinal (thymic) mass is present in 8 to 10 percent of childhood cases and in 15 percent of adult cases (Fig. 91–3). A bulky, anterior mediastinal mass can compress the great vessels and trachea and possibly lead to the superior vena cava syndrome. Patients with this syndrome present with cough, dyspnea, orthopnea, stridor, cyanosis, dysphagia, facial edema, increased intracranial pressure, and sometimes syncope. Painless enlargement of the scrotum can be a sign of testicular leukemia cell infiltration or hydrocele, the latter resulting from lymphatic obstruction. Both conditions can be readily diagnosed by ultrasonography. Overt testicular disease is relatively rare, is generally seen in infants or adolescents with T-cell leukemia and/or hyperleukocytosis, and does not require radiation therapy.71 Other uncommon presenting features include ocular involvement (leukemic infiltration of the orbit, optic nerve, retina, iris, cornea, or conjunctiva), subcutaneous nodules (leukemia cutis), enlarged salivary glands (Mikulicz syndrome), cranial nerve palsy, and priapism (resulting from leukostasis of the corpora cavernosa and dorsal veins or sacral nerve involvement). Epidural spinal cord compression at presentation is a rare but serious finding that requires immediate treatment to prevent permanent paraparesis or paraplegia. In some pediatric patients, infiltration of tonsils, adenoids, appendix, or mesenteric lymph nodes leads to surgical intervention before leukemia is diagnosed.
LABORATORY FEATURES
Anemia, neutropenia, and thrombocytopenia are common in patients with newly diagnosed ALL. The severity reflects the degree of marrow replacement by leukemic lymphoblasts (Table 91–3).18,65,66,67 Presenting leukocyte counts range widely, from 0.1 to 1500 × 109/L (median: 10–12 × 109/L). Hyperleukocytosis (>100 × 109 white cells/L) occurs in 11 to 13 percent of white children, but occurs more often in black children (23 percent) and in adults (16 percent) because they are more likely to have T-cell ALL. Profound neutropenia (<0.5 × 109/L) is found in 20 to 40 percent of patients, elevating their risk for infection. Approximately 90 percent of patients have circulating leukemic blast cells at diagnosis. Hypereosinophilia, generally reactive, may precede the diagnosis of ALL by several months. Some patients, mostly male, have ALL with the t(5;14)(q31;q32) chromosomal abnormality and a hypereosinophilic syndrome (pulmonary infiltration, cardiomegaly, and congestive heart failure). These patients often do not have circulating leukemic blasts or other cytopenias and have a relatively low percentage of blasts in the marrow.72 Activation of the interleukin-3 gene on chromosome 5 by the enhancer element of the immunoglobulin heavy-chain gene on chromosome 14 is thought to contribute to leukemogenesis and the associated eosinophilia in these cases.72 In patients with anemia, hemoglobin levels are lower in younger children. Occasionally, a child with ALL has a hemoglobin level as low as 1 g/dL.
| Percent of Total | ||
|---|---|---|
| Feature | Children, White/Black (%) | Adults (%) |
| Cell lineage | ||
| T cell | 15/24 | 25 |
| B-cell precursor | 85/76 | 75 |
| Leukocyte count (× 109/L) | ||
| <10 | 47–49/34 | 41 |
| 10–49 | 28–31/29 | 31 |
| 50–99 | 8–12/14 | 12 |
| >100 | 11–13/23 | 16 |
| Hemoglobin concentration (g/dL) | ||
| <8 | 48/58 | 28 |
| 8–10 | 24/22 | 26 |
| >10 | 28/20 | 46 |
| Platelet count (× 109/L) | ||
| <50 | 46/40 | 52 |
| 50–100 | 23/20 | 22 |
| >100 | 31/40 | 26 |
| CNS status* | ||
| CNS1 | 67–79/60 | 92–95 |
| CNS2 | 5–24/27 | ? |
| CNS3 | 3/3 | 5–8 |
| Traumatic lumbar puncture with blasts | 6–7/10 | ? |
| Leukemic blasts in marrow (%) | ||
| <90 | 33/46 | 29 |
| >90 | 67/54 | 71 |
| Leukemic blasts in blood | ||
| Present | 87/90 | 92 |
| Absent | 13/10 | 8 |
Decreased platelet counts are common at diagnosis (median: 48–52 × 109/L). Severe bleeding is uncommon, even when platelet counts are as low as 20 × 109/L, provided infection and fever are absent.73,74 Occasional patients, principally male, present with thrombocytosis (>400 × 109/L). Pancytopenia followed by a period of spontaneous hematopoietic recovery may precede the diagnosis of ALL in rare cases.75 Coagulopathy, usually mild, can be seen in 3 to 5 percent of patients, most of whom have T-cell ALL, and is only rarely associated with clinical bleeding.76 The level of serum lactate dehydrogenase is increased in most patients with ALL and is well correlated with tumor burden. Increased levels of serum uric acid are common in patients with a large leukemia cell burden, reflecting an increased rate of purine catabolism. Leukemic infiltration of the kidneys can lead to increased levels of creatinine, urea nitrogen, uric acid, and phosphorus. Rarely, patients present with hypercalcemia resulting from release of parathyroid hormone-like protein from lymphoblasts and leukemic infiltration of bone. The t(17;19)(q22;p13.3) with E2A-HLF fusion, found in 0.5 percent of B-cell precursor ALL, is associated with adolescent age group, disseminated coagulopathy, hypercalcemia, and dismal prognosis.77 Liver dysfunction as a result of leukemic infiltration occurs in 10 to 20 percent of patients and is usually mild. However, recognition of carriers of hepatitis B virus is important because prompt lamivudine therapy can prevent serious complications from virus reactivation during immunosuppressive treatment.78
Chest radiography is needed to detect enlargement of the thymus or mediastinal nodes and pleural effusions (see Fig. 91–3). Although bony abnormalities, such as metaphyseal banding, periosteal reactions, osteolysis, osteosclerosis, and osteopenia, can be found in 50 percent of patients, especially children with low leukocyte counts at presentation, skeletal films are not necessary for management. Magnetic resonance imaging (MRI) is useful in patients with suspected vertebral collapse or meningeal or nerve root involvement.
Examination of the cerebrospinal fluid (CSF) is an essential diagnostic procedure. Leukemic blasts can be identified in as many as one-third of pediatric patients and approximately 5 percent of adult patients at diagnosis of ALL; most of these patients lack neurologic symptoms.79,80 Traditionally, CNS leukemia is defined by the presence of at least 5 leukocytes/μL of CSF (with leukemic blast cells apparent in a cytocentrifuged sample) or by the presence of cranial nerve palsies. However, with the omission of prophylactic cranial irradiation in contemporary clinical trials, the presence of any leukemic blast cells in the CSF is associated with increased risk of CNS relapse and is an indication to intensify intrathecal therapy.80 Different opinions exist regarding when the first lumbar puncture should be performed. Most protocols now require the procedure at diagnosis and instill the first dose of chemotherapeutic agents intrathecally. Contamination of the CSF by leukemia cells as a result of traumatic lumbar puncture at diagnosis is associated with an inferior treatment outcome in children with ALL.81 The risk of traumatic lumbar puncture can be decreased by administering platelet transfusions to thrombocytopenic patients and by having the most experienced clinician perform the procedure.
Examination of a marrow aspirate is recommended for diagnosis of ALL because as many as 10 percent of patients lack circulating blasts at the time of diagnosis and because high concentrations of marrow cells are better than blood cells for genetic studies. Fibrosis or tightly packed marrow can lead to difficulties with marrow aspiration that necessitate biopsy. In patients with marrow necrosis, multiple marrow aspirations are sometimes needed to obtain diagnostic tissue.
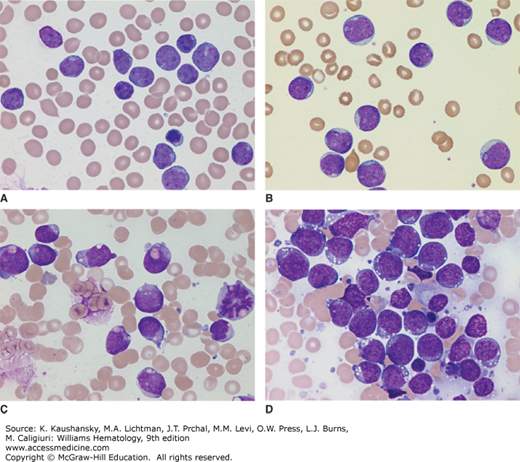
Diagnosis of ALL begins with morphologic analysis of Romanowsky-stained (Wright-Giemsa or May-Grünwald-Giemsa) marrow films. Lymphoblasts tend to be relatively small (ranging from the same size to twice the size of small lymphocytes) with scanty, often light-blue cytoplasm; a round or slightly indented nucleus; fine to slightly coarse and clumped chromatin; and inconspicuous nucleoli (Fig. 91–4A). In some cases, the lymphoblasts are large, with prominent nucleoli, moderate amounts of cytoplasm, admixed with smaller blasts (Fig. 91–4B). Cytoplasmic granules are found in the lymphoblasts of some patients with ALL (Fig. 91–4C). The granules usually are amphophilic (and stain fuchsia), readily distinguishable from primary myeloid granules (which stain deep purple), and demonstrated to be mitochondria by electron microscopy. B-cell blasts in Burkitt-type ALL are characterized by intensely basophilic cytoplasm, prominent nucleoli, and cytoplasmic vacuolation (Fig. 91–4D).
Figure 91–4.
A. Typical lymphoblasts with scanty cytoplasm, regular nuclear shape, fine chromatin, and indistinct nucleoli. B. Acute lymphoblastic leukemia (ALL) with large blasts showing prominent nucleoli, moderate amounts of cytoplasm, and an admixture of smaller blasts. C. ALL with cytoplasmic granules. Fuchsia granules are present in the cytoplasm of many blasts. Such granules may lead to a misdiagnosis of acute myeloid leukemia; however, the granules are negative for myeloperoxidase and myeloid-pattern Sudan black B staining. D. B-cell ALL lymphoblasts. The blasts in this phenotype are characterized by intensely basophilic cytoplasm, regular cellular features, and cytoplasmic vacuolation. (Images A to D, Wright-Giemsa stain; original magnification ×1000.)
Cytochemical stains (Sudan black stain and the stains for myeloperoxidase and the nonspecific esterases) distinguish ALL from acute myeloid leukemia but are now less commonly used for diagnosis than immunophenotyping.
Immunophenotyping is an essential part of the diagnostic evaluation. Antibodies distinguish clusters of differentiation (CD) groups, but most leukocyte antigens lack specificity. Hence, a panel of antibodies is needed to establish the diagnosis and to distinguish among the different immunologic subclasses of leukemic cells. Typical panels include antibodies to at least one highly sensitive marker (CD19 for B-cell lineage, CD7 for T-cell lineage, and CD13 or CD33 for myeloid cells) and antibodies to a highly specific marker (cytoplasmic CD79a and CD22 for B-cell lineage, cytoplasmic CD3 for T-cell lineage, and cytoplasmic myeloperoxidase for myeloid cells).17 Although ALL can be further subclassified according to the recognized steps of normal maturation within the B-cell lineage (pro-B, early pre-B, pre-B, transitional pre-B, and mature B cells) or T-cell lineage (pre-T, mid-, and late thymocyte) pathways, the only distinctions of therapeutic importance at present are those between T-cell, mature B, and other B-cell lineage (B-cell precursor type) immunophenotypes (Table 91–4).17 A distinct subset of T-cell ALL that retain stem cell–like features, termed early T-cell precursor ALL, has been identified and associated with a dire prognosis with conventional chemotherapy.82 Knowing the pattern of antigen expression at diagnosis is critically important for the detection of minimal residual disease by flow cytometry after treatment.83
| Subtype | Typical Markers | Childhood (%) | Adult (%) | Associated Features |
|---|---|---|---|---|
| B-cell precursor | CD19+, CD22+, CD79a+, cIg+/–, sIgμ–, HLA-DR+ | |||
| Pro-B | CD10– | 5 | 11 | Infants and adult age group, high leukocyte count, initial CNS leukemia, pseudodiploidy, MLL rearrangement, unfavorable prognosis |
| Early pre-B | CD10+ | 63 | 52 | Favorable age group (1–9 years), low leukocyte count, hyperdiploidy (>50 chromosomes) |
| Pre-B | CD10+/–, cIg+ | 16 | 9 | High leukocyte count, black race, pseudodiploidy |
| Mature B cell (Burkitt) | CD19+, CD22+, CD79a+, cIg+, sIg+ (kappa or lambda+) | 3 | 4 | Male predominance, initial CNS leukemia, abdominal masses, often renal involvement |
| T lineage | CD7+, cCD3+ | |||
| T cell | CD2+, CD1+/–, CD4+/–, CD8+/–, HLA-DR–, TdT+/– | 10 | 18 | Male predominance, hyperleukocytosis, extramedullary disease |
| Pre-T | CD2–, CD1–, CD4–, CD8–, HLA-DR+/–, TdT+ | 1 | 6 | Male predominance, hyperleukocytosis, extramedullary disease, unfavorable prognosis |
| Early T-cell precursor | CD1–, CD8–, CD5weak, CD13+, CD33+, CD11b+, CD117+, CD65+, HLA-DR+ | 2 | ? | Male predominance, age >10 years, poor prognosis |
Myeloid-associated antigens may be aberrantly expressed on otherwise typical lymphoblasts. Because of differences in monoclonal antibodies and immunophenotyping techniques, the frequencies of myeloid-associated antigen expression range from 5 to 30 percent in childhood cases and from 10 to 50 percent in adult cases.67,84 The pattern of myeloid-associated antigen expression is correlated with certain genetic features of blast cells. CD15, CD33, and CD65 are expressed in ALL cases with a rearranged MLL gene, and CD13 and CD33 are expressed in cases with the ETV6-RUNX1.84 There is a subset of cases that coexpress both lymphoid and myeloid markers but do not cluster with T-cell, B-cell precursor, or acute myeloid leukemia in gene-expression profiling. These cases may not respond to myeloid-directed therapy but attain remission with ALL-directed induction treatment.85,86 The presence of myeloid-associated antigens lacks prognostic significance in contemporary treatment programs but can be useful in immunologic monitoring of patients for minimal residual leukemia.67,83
ALL arises from a lymphoid progenitor cell that has sustained multiple specific genetic alterations that lead to malignant transformation and proliferation. Thus, genetic classification of ALL yields more relevant biologic information than any other means. Approximately 75 percent of adult and childhood cases can be readily classified into prognostically or therapeutically relevant subgroups based on the modal chromosome number (or DNA content estimated by flow cytometry), specific chromosomal rearrangements, and molecular genetic changes.1,2,17,18,19,20,41,54,55,56,87,88,89 Table 91–5 summarizes the prominent clinical and biologic features of cases with the most common genetic abnormalities.
| Subtype | Associated Features | Estimated Event-Free Survival (%) | |
|---|---|---|---|
| Children | Adults | ||
| Hyperdiploidy (>50 chromosomes) | Predominant precursor B-cell phenotype; low leukocyte count; favorable age group (1–9 years) and prognosis in children | 80–90 at 5 years | 30–50 at 5 years |
| Hypodiploidy (<45 chromosomes) | Predominant precursor B-cell phenotype; increased leukocyte count; poor prognosis | 30–40 at 3 years | 10–20 at 3 years |
| t(12;21)(p13;q22) [ETV6-RUNX1] | CD13+/–CD33+/– precursor B-cell phenotype; pseudodiploidy; age 1–9 years; favorable prognosis | 90–95 at 5 years | Unknown |
| t(1;19)(q23;p13.3) [TCF3-PBX1] | CD10+/–CD20–CD34– pre-B phenotype; pseudodiploidy; increased leukocyte count; black race; CNS leukemia; prognosis depends on treatment | 82–90 at 5 years | 20–40 at 3 years |
| t(9;22)(q34;q11.2) [BCR-ABL1] | Predominant precursor B-cell phenotype; older age; increased leukocyte count; myeloid antigens; improved early outcome with tyrosine kinase inhibitor treatment | 80–90 at 3 years | ~60 at 1 year |
| t(4;11)(q21;23) with MLL-AF4 fusion | CD10+/–CD15+/–CD33+/–CD65+/– precursor B-cell phenotype; infant and older adult age groups; hyperleukocytosis; CNS leukemia; poor outcome | 32–40 at 5 years | 10–20 at 3 years |
| t(8;14)(q24;q32.3) | Mature B-cell phenotype; L3 morphology; male predominance; bulky extramedullary disease; favorable prognosis with short-term intensive chemotherapy including high-dose methotrexate, cytarabine, and cyclophosphamide/ifosfamide | 75–85 at 5 years | 70–80 at 4 years |
| NOTCH 1 mutations | T-cell phenotype; favorable prognosis | 90 at 5 years | 50 at 4 years |
| HOX11 overexpression | CD10+ T-cell phenotype; favorable prognosis with chemotherapy alone | 90 at 5 years | 80 at 3 years |
| Intrachromosomal amplification of chromosome 21 | Precursor B-cell phenotype; low white blood cell count; intensified treatment required to avert a poor prognosis | 30 at 5 years | ? |
Two ploidy groups (hyperdiploidy >50 chromosomes and hypodiploidy <44 chromosomes) have clinical relevance. Hyperdiploidy, which is seen in approximately 25 percent of childhood cases and in 6 to 7 percent of adult cases, is associated with a favorable prognosis that may reflect an increased cellular accumulation of methotrexate and its polyglutamates, an increased sensitivity to therapeutic antimetabolites, and a marked propensity of these cells to undergo apoptosis.90,91 By contrast, hypodiploidy is associated with an exceptionally poor prognosis.88,89,92 Flow cytometric determination of cellular DNA content is a useful adjunct to cytogenetic analysis because it is automated, rapid, and inexpensive, and its measurements are not affected by the mitotic index of the cell population; results can be obtained in almost all cases. Flow cytometric studies can sometimes identify a small but drug-resistant subpopulation of near-haploid cells that may be missed by standard cytogenetic analysis.
Reciprocal chromosomal translocations in cases of B-cell and T-cell ALL arise from errors in the normal recombination mechanisms that generate antigen receptor genes. Such rearrangements can fuse the promoter/enhancer element of the immunoglobulin heavy- or light-chain gene or the T-cell antigen receptor β/γ or α/δ gene to sites adjacent to a variety of transcription factor genes. More often, genetic rearrangements result from the fusion of two genes encoding different transcription factors. These chimeric genes encode active kinases and altered transcription factors that regulate genes involved in the differentiation, self-renewal, proliferation, and drug resistance of hematopoietic stem cells.
Specific cytogenetic findings are correlated with presenting clinical features, blast-cell phenotypes, and clinical outcome (see Table 91–5). However, there are now compelling reasons to focus on molecular genetic lesions. First, molecular analyses can identify important submicroscopic genetic alterations not visible by standard karyotyping, such as the ETV6-RUNX1 fusion, intrachromosomal amplification of chromosome 21, deletions of tumor-suppressor genes, and mutations of protooncogenes.1,2,87,93,94,95 Second, cases with clinically important genetic rearrangements can be missed because of technical errors (e.g., karyotyping residual normal metaphase cells rather than leukemic metaphase cells). Hence, FISH and reverse transcriptase polymerase chain reaction (RT-PCR) assays are increasingly used. The application of microarray-based genome-wide analysis of gene expression and DNA copy number, complemented by transcriptional profiling, resequencing and epigenetic approaches, and next-generation sequencing have identified specific genetic alterations with biologic and therapeutic implications.
DIFFERENTIAL DIAGNOSIS
The initial manifestations of ALL can mimic a variety of disorders. The acute onset of petechiae, ecchymoses, and bleeding can suggest idiopathic thrombocytopenic purpura. The latter disorder often is associated with a recent viral infection, large platelets in blood films, normal hemoglobin concentration, and absence of leukocyte abnormalities in blood or marrow. Patients with ALL, or promyelocytic leukemia, or aplastic anemia can present with pancytopenia and complications associated with marrow failure. However, in aplastic anemia, hepatosplenomegaly and lymphadenopathy are rare, and the skeletal pain associated with ALL is absent. The results of marrow aspiration or biopsy usually distinguish between these diseases, although the diagnosis can be difficult in a patient who has hypocellular marrow that is later replaced by lymphoblasts. In one study, transient pancytopenia preceded ALL in 2 percent of all pediatric cases.75 During the preleukemic phase in these patients, polymerase chain reaction (PCR) analysis demonstrated monoclonality, suggesting inhibition of normal hematopoiesis by leukemia cells.96 ALL should be considered in the differential diagnosis of patients with hypereosinophilia, which can be a presenting feature of leukemia or can precede its diagnosis by several months. Occasionally, hematogones in a regenerative marrow may mimic leukemic blast cells; flow cytometry with optimal combinations of antibodies may be required to distinguish them.97
Infectious mononucleosis and other viral infections, especially those associated with thrombocytopenia or hemolytic anemia, can be confused with leukemia. Detection of reactive lymphocytes or serologic evidence of Epstein-Barr virus infection helps establish the diagnosis. Patients with acute infectious lymphocytosis, pertussis, or parapertussis can have marked lymphocytosis. However, even when leukocyte counts are as high as 50 × 109/L, the affected cells are mature lymphocytes rather than lymphoblasts. Bone pain, arthralgia, and occasionally arthritis mimic juvenile rheumatoid arthritis, rheumatic fever, other collagen diseases, or osteomyelitis. The marrow should be examined if glucocorticoid treatment is planned for presumed rheumatoid diseases.
In children, ALL should be distinguished from small, round cell tumors involving the marrow, including neuroblastoma, rhabdomyosarcoma, and retinoblastoma. Generally, in patients with solid tumors, a primary lesion will be found by standard diagnostic studies. Disseminated tumor cells often present in characteristic aggregates, and immunophenotypic characteristics of lymphoblasts are absent.
THERAPY
Optimal management of patients with ALL requires careful attention to supportive care, including immediate treatment or prevention of metabolic and infectious complications (Chap. 24) and rational use of blood products (Chaps. 138 and 139). Other important supportive care measures, such as use of indwelling catheters, amelioration of nausea and vomiting, pain control, and continuous psychosocial support for the patient and family, are essential.
Hyperuricemia and hyperphosphatemia with secondary hypocalcemia are frequently encountered at diagnosis, even before chemotherapy is initiated, especially in patients with B-cell or T-cell ALL or precursor B-cell leukemia with high leukemic cell burden.98 Patients should be given intravenous fluids; allopurinol or rasburicase (recombinant urate oxidase) to treat hyperuricemia; and a phosphate binder, such as aluminum hydroxide, calcium carbonate (if the serum calcium concentration is low), lanthanum carbonate, or sevelamer to treat hyperphosphatemia. Allopurinol, a relatively inexpensive drug, is usually used if the uric acid is less than 7 mg/dL. Allergic skin reactions occur in approximately 10 percent, and allopurinol should be stopped as soon as the risk of hyperuricemia from the destruction of a large leukemic cell burden has passed. By inhibiting de novo purine synthesis in leukemic blast cells, allopurinol can reduce the peripheral blast-cell count before chemotherapy.99 Allopurinol can decrease both the anabolism and catabolism of mercaptopurine by depleting intracellular phosphoribosyl pyrophosphate and by inhibiting xanthine oxidase. If mercaptopurine and allopurinol are given together orally, the dosage of mercaptopurine must be reduced.
Rasburicase works very rapidly and is extremely effective, especially for very elevated uric acid levels (>7 mg/dL), often with one infusion (a far smaller dose than the manufacturer recommends). Rasburicase breaks down uric acid to allantoin, a readily excreted metabolite that is five to 10 times more soluble than uric acid. Rasburicase is more effective than allopurinol, and it facilitates phosphorus excretion, partly because of rasburicase’s potent uricolytic effect (which obviates the need to alkalinize urine) and partly because of improved renal function with its use.100
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


