SELECTED EXAMPLES OF CYTOGENETIC AND MOLECULAR ABNORMALITIES IN LEUKEMIA



As with CBF fusions, the PML/RAR-α fusion protein functions as a dominant inhibitory oncogene for RAR-α–interacting proteins, including RXR-α. In addition, the PML/RAR-α fusion interferes with the function of the native PML protein, which is thought to function as a tumor suppressor gene.32 Collectively, the dominant interfering activities of the PML/RAR-α fusion protein result in a block in differentiation at the promyelocyte stage of development. The clinical response of these patients to ATRA is explained by the ability of this retinoid to bind to the PML/RAR-α fusion protein and reverse repression of target genes required for normal hematopoietic development. The ability of the PML/RAR-α fusion protein to repress transcription is partly due to the aberrant recruitment of the nuclear corepressor complex, including histone deacetylase, suggesting that pharmacologic agents that inhibit histone deacetylases may be useful in therapy of APL.24,33
The transforming properties of the PML/RARα fusion gene have been tested in murine models. Expression of PML/RAR-α in transgenic mice from promoters that direct expression to the promyelocyte compartment result in an APL-like phenotype.34–36 However, there is approximately a 6-month lag before the development of leukemia, incomplete penetrance of approximately 15% to 30%, and acquired karyotypic abnormalities, all suggesting that second mutations are required for induction of leukemia. In at least some cases, activating mutations in FLT3, as discussed later in “Activating Mutations in FLT3 and KIT,” may be the additional mutation required. ATRA is efficacious in leukemic animals, expressing both PML/RAR-α and activated FLT3, and this model has allowed for the preclinical testing of novel agents such as arsenic trioxide.37
CHROMOSOMAL TRANSLOCATIONS THAT TARGET HOX FAMILY MEMBERS
The large HOX family of transcription factors is important in patterning in vertebrate development and also plays a critical role in normal hematopoietic development (reviewed in ref. 38). HOX genes may also be targeted by chromosomal translocations, with examples including the NUP98/HOXA9 and NUP98/HOXD13 fusions, associated with t(7;11) and t(2;11), respectively (see Table 31.1).39,40 HOX gene expression is tightly regulated during hematopoietic development. HOXA9, for example, is expressed in early hematopoietic progenitor cells but is down-regulated during hematopoietic differentiation and is undetectable in terminally differentiated cells. It has been suggested that unregulated overexpression of the HOXA9 moiety from the constitutively active NUP98 promoter may result in aberrant differentiation. Experimental support for this hypothesis includes the observation that the NUP98/HOXA9 fusion protein can transform 3T3 fibroblasts, an activity that requires the HOXA9 DNA-binding domain.41
The contribution of the NUP98 moiety to leukemic transformation is not fully understood. NUP98 is normally a component of the nuclear pore complex and is constitutively and ubiquitously expressed. However, several lines of evidence suggest that NUP98 contributes more than a constitutively activated promoter. For example, NUP98 motifs known as FG repeats are essential for transformation and may serve to recruit transcriptional coactivators, such as CBP/p300, to HOXA9 DNA-binding sites.41 In murine models of leukemia, overexpression of HOXA9 alone is not sufficient to cause AML, but coexpression of HOXA9 with transcriptional cofactors, such as MEIS1, results in efficient induction of AML. Thus, the NUP98 moiety in the context of the NUP98/HOXA9 fusion may serve multiple functions, including provision of an active promoter, and recruitment of transcriptional coactivators such as CBP/p300 that subserve the function of other cofactors such as MEIS1. Epidemiologic evidence that the NUP98 moiety contributes to leukemogenesis includes the observation that there are now a spectrum of fusion proteins involving components of the nuclear pore that are targeted by chromosomal translocations in acute leukemias. These include NUP98 and NUP214 fused to a diverse group of partners, including HOXA9 and HOXD13, and the DDX10, PMX1, DEK, and ABL1 genes, respectively.
It has been hypothesized that dysregulated HOX gene expression may be important in leukemias that do not directly target HOX family members. Several proteins that are upstream of HOX expression have been observed as fusion genes associated with AML, the most frequent of these are MLL gene rearrangements. More than 40 chromosomal translocations target MLL and result in fusions of MLL with a broad spectrum of partners. However, a common biologic feature of all of these may be their ability to dysregulate HOX gene expression during hematopoietic development. For example, t(12;13) associated with AML results in expression of high levels of CDX2 from the TEL locus.42 CDX2 is a homeotic protein that regulates expression of HOX family members in the colonic epithelium. As in hematopoietic development, HOX gene expression is highest in colonic stem cells in the colonic crypts and is down-regulated with maturation. It has been shown that CDX2 and CDX4 can dysregulate HOX expression in hematopoietic progenitors and result in leukemia.42,43 Evidence to support this includes the ability of CDX2 to induce leukemia in murine retroviral transduction models.42,43 Although CDX2 is not normally expressed in hematopoietic cells, a family member, CDX4, has been cloned and appears to play a similar role in hematopoietic development as CDX2 does in the gut. Of note, CDX4 in hematopoietic cells appears to either be downstream or epistatic with MLL in regulation of HOX gene expression.44
Taken together, these data indicate that the NUP98/HOXA9 fusion transforms hematopoietic progenitors in part through dysregulated overexpression and by transactivation mediated through the NUP98 transactivation domain that recruits CBP. However, like other gene rearrangements involving hematopoietic transcription factors, expression of NUP98/HOXA9 alone is not sufficient to cause leukemia. In murine bone marrow transplant models, NUP98/HOXA9 induces AML only after markedly prolonged latencies indicative of a requirement for second mutation.
CHROMOSOMAL TRANSLOCATIONS THAT TARGET THE MLL GENE
As noted earlier in “Recurring Chromosomal Abnormalities in Acute Leukemia,” the MLL locus is involved in more than 40 different chromosomal translocations with a remarkably diverse group of fusion partners,45,46 and are associated with mostly FAB subtype M4 or M5, and fewer with M2 AML. Patients who have received prior chemotherapy for cancer and develop AML (therapy-related myelodysplastic syndrome [MDS]/AML, t-AML) often have abnormalities in 11q23, especially those patients treated with topoisomerase inhibitors such as etoposide or topotecan. Chromosomal translocations involving band 11q23 result in expression of a fusion gene containing amino-terminal MLL sequences fused to a wide variety of partners. There has been no common functional motif or activity ascribed to all partners; however, specific fusions may be associated with specific leukemic phenotypes. The MLL/AF4 fusion associated with t(4;11) is frequently observed in infant leukemias and is associated with an ALL phenotype in more than 90% of cases, whereas the MLL/AF9 fusion associated with the t(9;11) is almost exclusively associated with AML. Certain MLL fusion genes also have prognostic significance. For example, patients with t(9;11)(p22;q23) have a better outcome than those with other translocations involving 11q23.
The MLL gene encodes a large, ubiquitously expressed protein. The Drosophila protein trithorax, a homologue of MLL, regulates patterning and HOX gene expression during development. It has been hypothesized, in part based on these observations, that MLL might be required for maintenance of HOX gene expression. In support of this hypothesis, mice that lack Mll express HoxA7 but are not able to maintain its expression.47 Mice that have homozygous deficiency for Mll have an embryonic lethal phenotype at day 10.5 postconception. Even heterozygous animals have developmental anomalies in the axial skeleton and hematopoietic deficits including anemia.47 Thus, as for other genes targeted by chromosomal translocations, MLL is important for normal hematopoietic development.
The function of MLL is not fully understood, but cell culture and murine models have provided some insight into transforming activity of the fusion proteins. The MLL protein of the fusion protein retains the amino terminal AT hooks that facilitate binding to DNA, as well as a methyltransferase domain. With the exception of CBP/p300, the function of the remaining broad spectrum of divergent fusion partners is poorly understood. In fact, the remarkable divergence of partners has suggested that alteration in the MLL gene itself is a critical required event for transformation. In support of a central role of MLL rearrangement in AML, it has been reported that partial tandem duplications of MLL are associated with AML, in particular AML associated with +11.48 Data demonstrating that MLL is a processed polypeptide provide further support for this hypothesis. MLL is processed by proteolytic cleavage into two component parts by a novel protease called taspase 1.49 Cleavage is required for normal regulation of expression of anterior and posterior HOX gene paralogs during development. It has thus been suggested that MLL fusion genes, which are not cleavable, may mimic the uncleaved native MLL protein, thereby dysregulating HOX gene expression.44
MLL fusion genes have transforming properties in serial replating assays in retrovirally transduced hematopoietic progenitors as well as in murine models (reviewed in ref. 45). Although various MLL fusions have similar transforming properties in vitro, there are distinctive differences in disease penetrance and latency in the murine models, depending on the fusion partner. It is possible that the MLL gene rearrangement may be critical for transformation, whereas the fusion partners confer properties related to disease phenotype. The long latency of disease in murine models supports the hypothesis that MLL fusions, like the PML/RAR-α and CBF-related fusion proteins, require second mutations to cause leukemia.
As noted in “Leukemic Stem Cell,” data indicate that certain MLL fusion genes may also confer properties of self-renewal to hematopoietic progenitors. MLL/ENL expression in common myeloid progenitors or granulocyte-monocyte progenitors in a murine system conferred properties of self-renewal, including the ability to serially replate in methylcellulose cultures and to engender a transplantable AML phenotype in recipient animals.5 Similarly in a mouse model of MLL-AF9 oncogene-induced leukemia, up to a quarter of the leukemic cells exhibit stem cell behavior.7 Furthermore the MLL-AF9–positive leukemic stem cells are heterogeneous as they give rise to ALL when injected into immunodeficient mice. The same cells cause AML when injected into immunodeficient mice that are transgenic for the human genes SCF, GM-CSF, and IL-3.51 These data indicate that leukemogenic mutations may occur in cells that have no intrinsic self-renewal capacity and yet confer these properties by activation of specific transcriptional programs, which may be further modified by clues from microenvironment. These exciting observations provide tools for identification of target genes that confer properties of self-renewal and may have value as therapeutic targets for treatment of leukemia.
CHROMOSOMAL TRANSLOCATIONS THAT INVOLVE TRANSCRIPTIONAL COACTIVATORS AND CHROMATIN REMODELING PROTEINS
Several translocations associated with leukemia involve transcriptional coactivators and chromatin-modifying proteins that have no apparent DNA-binding specificity. These include the MLL/CBP and MOZ/CBP fusions that involve the transcriptional coactivator CBP and the MLL/p300 and MOZ/TIF2 fusions, which involve the coactivators p300 and TIF2, respectively.52,53 Although TIF2 itself is not known to have histone acetylase transferase (HAT) activity, a hallmark of the coactivators CBP and p300, it has a well-characterized CBP interaction domain that serves to recruit CBP into a complex with MOZ/TIF2.54 Thus, recruitment of CBP/p300 is a shared theme among this group of fusion genes.
The transcriptional targets and transformation properties of this class of fusion proteins are not fully understood. Transduction of MLL/CBP into primary murine bone marrow cells followed by transplantation results in a long-latency AML, suggesting the need for secondary mutations.55 MOZ/TIF2 also results in leukemia in a similar model system. MOZ is a HAT protein that contains a nucleosome-binding domain and an acetyl–coenzyme A–binding catalytic domain. Mutational analysis shows that leukemogenic activity requires MOZ nucleosome-binding activity and CBP recruitment activity, but the MOZ HAT activity is dispensable. These data would be consistent with a CBP gain of function in which CBP is recruited to MOZ nucleosome-binding sites.54 However, it has also been hypothesized that the leukemogenic potential of this class of fusions may be related to dominant negative interference with CBP/p300 or that the translocation leads to simple loss of function of CBP expressed from one allele. In support of this hypothesis, loss of a single allele of CBP/p300 in the human Rubinstein-Taybi syndrome increases predisposition to malignancies including colon cancer, and mice that are heterozygous for CBP that develop hematopoietic tumors.56
t(1;22) TRANSLOCATION ASSOCIATED WITH INFANT ACUTE MEGAKARYOBLASTIC LEUKEMIA
Until recently, little was known about the molecular pathogenesis of acute megakaryoblastic leukemias (AMKLs; FAB M7), partly because of the difficulty in obtaining adequate quantities of material for analysis from densely fibrotic bone marrow. The t(1;22) that is associated with the majority of non–Down syndrome AMKL in infants has been cloned. The translocation results in expression of the OTT1/MAL fusion gene.57,58 OTT1 (RBM15) contains three amino-terminal RNA recognition motifs and a Spen paralog and ortholog C terminal motif that is conserved in Drosophila. Ott1 deletion in mice reveals multiple roles in hematopoietic development including megakaryocyte growth.59 The MAL (MKL1) gene is a Rho-GTPase-regulated cofactor for serum response factor and controls megakaryocyte development.60,61 A knock-in mouse model expressing OTT1/MAL is able to recapitulate AMKL and demonstrated that constitutive transcriptional activation from RBPJk, a downstream Notch effector, is essential for its pathogenesis.62
DELETIONS AND NUMERIC ABNORMALITIES IN ACUTE LEUKEMIAS
Deletions of all or part of chromosomes 5 and 7 and trisomy 8 are among the most common chromosomal abnormalities associated with gain or loss of genetic material but are not associated with a specific subtype of AML. They are considerably more frequent in older patients, whereas the frequency of the specific translocations and inversions described previously decreases with age. These same abnormalities (+8, −7, −5/5q-) are more common in patients with an antecedent MDS, therapy-related AML, or exposure to environmental mutagens. Specific translocations or inversions are relatively less common in these patient groups. An exception to this is AML, which develops in patients who have received high doses of etoposide for treatment of a previous malignancy. As noted in “Chromosomal Translocations that Target the MLL Gene,” translocations involving 11q23 are commonly observed in this setting.
The high frequency of deletions of the long arm of chromosome 5 (5q-) has interest because the genes encoding several hematopoietic growth factors and their receptors are located on this arm, including GM-CSF (5q23–31), IL-3 (5q23–31), and IL-4 (5q23–31). The M-CSF receptor, c-fms, and the PDGFβR are also on this chromosome, at 5q33. In some 5q- chromosome defects, one or more of these genes may be deleted. An intensive effort has been made to identify putative tumor suppressor genes in several critically deleted regions of chromosome 5q, as well as 7q and 20q (reviewed in ref. 63). In addition, genomic wide loss of heterozygosity screens have identified a spectrum of other smaller recurrent deletions associated with AML.64
An RNA-mediated interference (RNAi)-based screen of each gene within the common deleted region led to identification of RPS14 gene. A partial loss of function of the ribosomal subunit protein RPS14 in normal hematopoietic progenitor cells recapitulated the disease with impaired erythropoiesis and relative preservation of the other lineages. Conversely, a forced expression of RPS14 rescued the disease phenotype in patient-derived bone marrow cells. In patients with the 5q- syndrome, 1 allele of RPS14 is deleted, and haploinsufficient expression of RPS14 has been confirmed in patient samples. These results indicate that the RPS14 gene is a causal gene in the 5q- syndrome.65 However, it is conceivable that other genes (on 5q or elsewhere) collaborate with RPS14 to cause the disease phenotype and eventually to progress to AML. In that regard, the 5q- syndrome region on chromosome 5 should be distinguished from a more centromeric locus on 5q that has been associated with therapy-related and aggressive subtypes of MDS as well as AML, and for which two candidate genes, CTNNA1 and EGR1, have been recently reported.66,67
Acquired deletions are a hallmark of cancer and precancerous states. In general, such deletions flag the existence of a tumor suppressor gene conforming to two-hit hypothesis, in which one allele is often deleted and the other allele is inactivated by deletion, mutation, or epigenetic modification. However, the search for the key tumor suppressor gene has been elusive in the 7q or 20q deleted regions in MDS or AML. Several possible explanations can be made for the difficulty in identification of classic tumor suppressors, despite the availability of complete genomic sequence and detailed annotations of expressed sequences in these regions. The residual allele may be affected by epigenetic mutations that interfere with expression, such as promoter or aberrant methylation, or both, but do not affect coding sequence. These types of mutations are more difficult to detect. Alternatively, it is possible that haploinsufficiency for one or more genes in the critically deleted loci is responsible for the MDS/AML phenotype.68 Haploinsufficiency for the transcription factor AML1 has been reported in a familial leukemia syndrome, and haploid gene dosage is increasingly being identified as a genetic basis for inherited human diseases.69,70 RNAi-based discovery of the 5q- syndrome gene suggests that haploinsufficient disease genes can be identified with this approach.
CHROMOSOMAL TRANSLOCATIONS THAT RESULT IN OVEREXPRESSION OF OTHERWISE NORMAL GENES
The chromosomal translocations described thus far result in expression of aberrant fusion genes. Chromosomal translocations may also result in overexpression of otherwise normal genes as a result of juxtaposition of a gene not normally expressed in adult hematopoietic tissues adjacent to an active promoter or enhancer. Most of those identified thus far involve the immunoglobulin or T-cell receptor (TCR) enhancer loci, and thus most of these are associated with lymphoid malignancies.
The prototypical example of juxtaposition of an immunoglobulin enhancer locus to an oncogene resulting in B-cell leukemia and lymphoma is the t(8;14)(q24;q32), resulting in overexpression of the MYC bHLH/bZIP transcription factor on chromosome 8 because of juxtaposition to the immunoglobulin heavy-chain enhancer on chromosome 14. Similar phenotypes ensue from juxtaposition to other immunoglobulin enhancers in the human genome, such as the Igk locus on chromosome 2 or the Igl locus on chromosome 22, and are characterized as B-ALL or lymphoma (Table 31.1). Overexpression of MYC from immunoglobulin enhancers in murine models results in B-cell leukemias and lymphomas, confirming a central role for MYC overexpression in transformation. However, the mechanism of transformation of MYC, and, indeed, a complete understanding of its target genes, is not fully understood. MYC is fully active as a transcription factor when heterodimerized with MAX. MAX is normally a homodimer, or a heterodimer complexed with MAD, which represses transcription. Overexpression of MYC is thought to shift the equilibrium in favor of an MYC-MAX homodimer that transactivates genes that confer the leukemic phenotype to B cells.
CHROMOSOMAL TRANSLOCATIONS INVOLVING THE T-CELL RECEPTOR
T-cell leukemias are often associated with overexpression of a number of genes because of juxtaposition to the TCR enhancer loci (TCRβ at chromosome 7q34 or TCRα/d at chromosome 14q11). Overexpression is thus associated with T-cell phenotypes, including T-cell ALL and lymphoma. For example, T-cell ALL may be associated with overexpression of bHLH family members that include TAL1/SCL, TAL2/SCL2, LYL1, HOX11, HOX11L2, LMO2, LMO1, and MYC (Table 31.1; reviewed in refs. 71 and 72). In addition to the minority of T-ALL cases with gene rearrangements involving these loci, it has been demonstrated that many patients without evident cytogenetic abnormalities overexpress TAL1, LMO2, HOX11, or HOX11L2.
POINT MUTATIONS IN ACUTE LEUKEMIA
Although intensive effort has focused on chromosomal translocations in leukemia, in part because of their high frequency in various kinds of leukemia, it has become increasingly clear that point mutations play an important role in a spectrum of leukemias (Table 31.2). Ongoing high-throughput sequencing initiatives have identified numerous solitary and recurring somatic point mutations within the various leukemic subtypes. The interpretation of these sequencing data requires the identification of “driver” versus “passenger” mutations. Driver mutations cause genetic alterations contributing to leukemic pathophysiology, whereas passenger mutations occur in leukemia cells and are propagated but are not etiologic to the disease.73 It is therefore essential that newly discovered somatic mutations in leukemia through sequencing studies undergo subsequent biologic validation in an experimental model system.
TABLE 31.2
POINT MUTATIONS IN ACUTE LEUKEMIA

Oncogenic RAS Mutations
Activating mutations in RAS may be associated with AML and MDS, typically at codons 12, 13, or 61, or N– or K-RAS. The reported incidence varies widely between studies from 25% to 44% and RAS mutations may confer a worse prognosis (reviewed in ref. 74). Considerable effort has been devoted to developing small-molecule inhibitors of RAS activation, with a focus on prenylation inhibitors, including farnesyl transferase and geranyl-geranylation inhibitors that preclude appropriate targeting of activated RAS to the plasma membrane.75 Specifically targeting activated RAS mutants remains an attractive option, and prenyltransferase inhibitors appear to have activity in AML. However, clinical activity is not correlated with the presence of activating mutations in RAS or even with inhibition of the target farnesyl transferase itself.75,76 Several possible interpretations can be made of these observations, including the possibility that RAS is activated by mechanisms other than intrinsic point mutations (e.g., constitutively activated tyrosine kinases such as FLT3), or that other proteins that are targets of prenylation are important in leukemia pathogenesis, or that farnesyl transferase inhibitors have off-target effects.
Activating Mutations in Tyrosine Kinases
One of the more exciting recent developments in the pathogenesis of AMLs has been identification and characterization of activating mutations in hematopoietic tyrosine kinases. Substantial evidence has been shown that chromosomal translocations that activate tyrosine kinases can contribute to the pathogenesis of CML syndromes. The most common of these is the BCR/ABL gene rearrangement, but other examples include the TEL/ABL, TEL/PDGFβR, TEL/JAK2, H4/PDGFβR, FIP1/PDGFβR, and rabaptin/PDGFβR fusion proteins. However, these fusion genes are only rarely encountered in AMLs. Approximately 1% to 2% of cases of de novo AML have the BCR/ABL gene rearrangement. In addition, there are very rare cases of disease progression from CML to AML associated with acquisition of second mutations such as the NUP98/HOXA9, AML1/ETO, or AML1/EVI1 rearrangement noted in Table 31.1. However, point mutations in the tyrosine kinase activation loop and juxtamembrane (JM) mutations that activate FLT3 and c-KIT have been identified in a significant proportion of AML cases. These findings may have important therapeutic implications with the demonstration of the efficacy of molecular targeting of the ABL kinase in BCR/ABL-positive CML and CML blast crisis with imatinib.77
Activating mutations in FLT3 have been reported in approximately 30% to 35% of cases of AML.78 In 20% to 25% of cases, internal tandem duplications (ITDs) of the JM domain result in constitutive activation of FLT3. These can range in size from a few to more than 50 amino acids and are always in frame. Because of the extensive variability in size and exact position of the repeats within the JM domain, it has been hypothesized that these are loss-of-function domains that impair an autoinhibitory domain, resulting in constitutive kinase activation in the absence of ligand. In support of this, the crystallographic structure of FLT3 demonstrates a 7 amino acid extension of the JM domain that intercalates into the catalytic domain, thereby precluding kinase activation.79 It is likely that ITD mutations in this region would disrupt structure of the autoinhibitory domain, resulting in kinase activation. In an additional 5% to 10% of cases, so-called activating loop mutations occur near position D835 in the tyrosine kinase.80 Several large studies have confirmed the frequency of these mutations in adult and pediatric AML populations and the fact that mutations in FLT3 appear to confer a poor prognosis.81–83 High-throughput sequencing of AML patient samples lacking known FLT3 mutations revealed nine novel acquired mutations resulting in amino acid changes within the extracellular, JM, and activation domains; however, only four of the nine changes were driver mutations capable of kinase activation and conferring growth factor independence, thus emphasizing the need for biologic validation of sequencing data.84
FLT3 mutations may occur in conjunction with known gene rearrangements, such as AML1/ETO, PML/RARα, CBFβMYH11, or MLL. Analogous activating loop mutations at position D816 have also been reported in C-KIT in approximately 5% of cases of AML. Activating mutations in the thrombopoietin receptor, MPLW515L, originally identified in myelofibrosis with myeloid metaplasia, and MPLT487A have been observed in both primary cases of AMKL and those secondary to myeloid metaplasia.85–87
The Janus kinase family (JAK1-3) of nonreceptor tyrosine kinases, in addition to involvement in translocation-derived fusions such as TEL/JAK2, have been found to contain activating point mutations. JAK kinases are important signaling intermediaries of multiple hematopoietic cytokine receptors and downstream effectors such as STAT proteins.88 JAKV617F, originally identified as a causative mutation in polycythemia vera, is also seen to a minor extent in AML.89 Additional mutations in JAK2 and JAK3 have been isolated in AMKL.87,90,91 Mutations in JAK1, 2, or 3 are found in approximately 11% of BCR/ABL-negative childhood acute lymphoid leukemia and were often concurrent with deletion of the IKAROS lymphoid-specific transcription factor and the CDKN2A/B tumor suppressor.92 As sequencing efforts continue, it is probable the list of activating kinase mutations will dramatically increase. As kinases are proving to be relatively amenable to targeted therapy, the opportunities for treatment tailored to these activated kinases should likewise expand.
MUTATION IN TUMOR SUPPRESSOR GENES
Wilms’ tumor gene was originally described as a tumor suppressor gene in patients with WAGR (Wilms’ tumor predisposition-aniridia-genitourinary-mental retardation).93 WT1 is found in adult tumors from different origin, and these tumors arise in tissues that normally do not express WT1. It has therefore been suggested that expression of WT1 might play an oncogenic role in these tumors (reviewed in ref. 94). WT1 is located at the chromosome 11p and encodes for a transcription factor with N-terminal transcriptional regulatory domain and C-terminal zinc finger domain (exon 7 to 10). The expression of WT1 inversely correlates with the degree of differentiation in the hematopoietic system as it is present in CD34+ cells and absent in mature leukocytes.94,95 WT1 functions as a potent transcription regulator of genes important for cell survival and cell differentiation. The disruption of WT1 function promotes stem cell proliferation and hampers differentiation.94 Although the precise role of WT1 in normal and malignant hematopoiesis remains to be further elucidated, it seems to have a dual role in leukemia.96
The wild type form of WT1 is highly (75% to 100%) expressed in a variety of acute leukemias.97 Consistent with the function of an oncogene is the pattern of WT1 expression in CML, where low levels are found in the chronic phase but are frequently increased in the accelerated and blast crisis phase.98 High levels of WT1 in patients after chemotherapy is associated with poor prognosis.
WT1 can act as a tumor suppressor in mice.99 Mutation of the WT1 gene can be detected in approximately 10% of normal karyotype AMLs.100,101 Mutations that cluster to exon 7 (mostly frameshift mutations resulting from insertions and deletions) and exon 9 (mostly substitutions) are associated with poor clinical outcome.101–103 These data are examples of WT1 as a tumor suppressor. On the other hand, a recent study has analyzed mutations within the entire WT1 coding sequence in a very large cohort of young adults with normal karyotype AML. Contrary to the previous observations,101–103 WT1 mutations had no prognostic impact.104 The different results from these large studies could be explained by the variable biological role of WT1 in AML, possible differences in therapy, and other patient characteristics. It is therefore desirable that testing for WT1 mutations becomes part of the risk assessment in future clinical trials to resolve these discrepancies.
TP53 is a tumor suppressor that induces cell-cycle arrest in a response to apoptotic cell death or DNA repair due to genotoxic substances, oncogenes, hypoxia, DNA damage, or ribonucleotide depletion.105 Inactivation of TP53 plays an important role during neoplastic transformation in solid tumors and also during progression of hematologic malignancies.106–108 Animal experiments suggest that the loss of one TP53 allele could be sufficient for tumorigenesis.109 This could be relevant for the development of leukemia in patients with single TP53 deletion. The loss of 17p in AML is often accompanied by a TP53 mutation resulting in a loss of heterozygosity.110,111 Another possibility is the inactivation of downstream mediators of TP53, which affect not only cell-cycle arrest but also DNA repair and apoptosis. Alternatively, overexpression of genes inhibiting or promoting degradation of TP53 can be considered; for example, MDM2 gene amplifications have been detected in B-CLL.122 TP53 deletion can be present as a loss of 17p as a part of a complex aberrant karyotype or as a single chromosomal aberration, both resulting in a poor clinical outcome.110,113–115
The incidence of TP53 aberrations is high in AML with a complex aberrant karyotype (up to 70%),110 but relatively rare in other AML groups (2% to 9%),83,110,116 and TP53 mutations without cytogenetic alteration are to rare events.111,117 Low-risk AML t(8;21) or inv(16) is not associated with TP53 deletion. There is significant positive association between TP53 deletion and other high-risk chromosomal aberrations such as del(5q), and monosomy 5 and 7.115,118 Molecular risk factors FLT3-ITD and NPM1 mutation do not seem to cluster with the TP53 deletion in complex karyotype patients.115 TP53-deleted cells have greater resistance to various conventional antileukemic drugs.119 Although published data of multidrug-resistance gene expression showed negative influence on therapy response in complex aberrant patients,120 the association of TP53 deletion and MDR1 expression has been confirmed for CML, but not for AML.121 Hence, an independent mechanism of resistance needs to be considered.115 Taken together, TP53 deletion is a high-risk factor conveying a poor outcome, and further studies are necessary to provide and evaluate alternative therapies.
ACTIVATING MUTATIONS OF NOTCH
NOTCH1 is a component of an evolutionarily conserved pathway shown to direct T-cell lineage determination in early and late stages of lymphocyte development as well as play a role in hematopoietic stem cell self-renewal (reviewed in ref. 122). NOTCH1 is a heterodimeric transmembrane receptor. Ligand binding to NOTCH1 allows proteolytic cleavage of the heterodimerization domain (HD) by γ-secretase of the C- terminal intracellular domain (ICN), which then localizes to the nucleus to function as a transactivator. Involvement of NOTCH1 in T-ALL had been observed with the rare t(7;9)(q34;q34.3) in which translocation of TCRβ locus into the NOTCH1 gene results in the expression of the truncated, transcriptionally active ICN.
Recently, a series of point mutations in NOTCH1 were identified in over half of all T-ALL cases123 (reviewed in ref. 124). These mutations clustered in two primary locations, the HD and the proline, glutamate, serine, and threonine-rich (PEST) domain. The missense mutations within the HD domain make NOTCH1 more amenable to γ-secretase–mediated cleavage, thus enhancing activation. The PEST domain controls the rate of degradation of the activated ICN. PEST domain mutants are primarily small insertions/deletions into the reading frame causing deletion of all or part of the domain and extending the half-life of the activated ICN. Fortuitously, γ-secretase inhibitors (GSIs) had already undergone significant clinical development from the involvement of γ-secretase in processing the pathogenic β-amyloid peptide associated with Alzheimer dementia. Initial clinical trials of GSIs in T-ALL have shown minimal effects on disease and significant gastrointestinal toxicity.125 Use of GSIs in combination with agents affecting alternative pathways may provide synergism and improve efficacy. Treatment of a mouse model of T-ALL with GSIs and corticosteroids has demonstrated that GSIs are capable of abrogating corticosteroid resistance in established cell lines as well as limiting GSI-mediated gut toxicity.126
MUTATIONS ALTERING LOCALIZATION OF NPM1
NPM1 (nucleophosmin1) encodes a protein that acts as a molecular chaperone between the nucleus and cytoplasm. It is involved in multiple cellular processes including regulation of TP53/ARF pathways, ribosome biogenesis, and duplication of centrosomes. NPM1 had been previously identified in acute leukemias as a translocation fusion partner with RAR and MLF as well as with ALK in anaplastic large cell lymphoma. Aberrant cytoplasmic localization of NPM1 has been observed in 25% to 30% of adult AML and is associated with point mutations within exon 12, which are hypothesized to enhance a nuclear export motif within the expressed protein.127 The mechanism by which mutated NPM1 causes leukemia is not clear; however, the cytoplasmic localization of NPM1 is thought to be intrinsic to its altered function.128 NPM1 mutations are found more frequently in AML with normal karyotypes (50% to 60%) and more apt to have FLT3-ITD mutations as well. Among normal cytogenetic AMLs, the presence of cytoplasmic NPM1 in the absence of the FLT3-ITD is associated with a more favorable prognosis.128
LOSS-OF-FUNCTION POINT MUTATIONS IN AML1,C/EBPα, AND GATA-1
AML1 (also known as RUNX1, CBFA2) is a frequent target of translocations in human leukemias. In addition to frequent involvement of AML1 as a consequence of chromosomal translocations, it has been determined that loss-of-function mutations in AML1 are responsible for the inherited leukemia syndrome FPD/AML (familial platelet disorder with propensity to develop acute myelogenous leukemia).69,129 In addition, approximately 3% to 5% of sporadic cases of AML harbor loss-of-function mutations in AML1,69,130 with a higher frequency in M0 AML (25%) and in AML or MDS with trisomy 21. It is not known whether loss-of-function mutations in AML1 confer the favorable prognosis associated with translocations involving the AML1 gene.
C/EBPα is a 42-kDa hematopoietic transcription factor that is required for normal myeloid lineage development. Because many translocations associated with AML phenotypes result in loss of function of hematopoietic transcription factors, it has been hypothesized that C/EBPα may also be a target for loss-of-function mutations in human leukemia. Two major types of C/EBPα point mutations have been described in AML: short frame-shifting mutations in the region encoding the amino-terminus causing expression of a shortened 30-kDa protein with dominant negative activity and in-frame insertions or deletions in the region of the carboxy-terminus that alter the DNA-binding or dimerization domains causing loss of function.131 Thus, these mutations would be predicted to impair hematopoietic differentiation. Although the bulk of C/EBPα occur in patients with normal cytogenetics, overall and progression-free survival is closer to the favorable rather than intermediate prognostic group.132 Therefore, C/EBPα mutational status may provide a more accurate risk assessment for normal cytogenetic AML.
GATA-1 mutations are associated with a subset of AMKLs (FAB M7), in particular leukemias arising in patients with Down syndrome (constitutional trisomy 21). These mutations result in early termination of the full-length GATA-1 protein; however, translation of a short form, GATA-1s, from an alternate initiation codon occurs. GATA-1s is theorized to function as either a hypomorphic or dominant negative allele and dysregulation of GATA-1 pathways is thought to contribute to leukemogenesis.133,134 GATA-1 mutations are often seen in a tansient myeloproliferative disorder that precedes Down syndrome–associated AMKL, suggesting that GATA-1 mutation is an early event.133 GATA-1 mutations thus far have only been associated with Down syndrome and have not been observed in other infant AMKLs, including those with the t(1;22) described earlier in “t(1;22) Translocation Associated with Infant Acute Megakaryoblastic Leukemia.”
MUTATION OF LYMPHOID DEVELOPMENT GENES IN ALL
Analysis of pediatric acute leukemia samples through a combination of high-resolution SNP arrays and genomic sequencing revealed numerous cryptic translocations, small deletions, and point mutations affecting genes required for B-cell commitment and differentiation.135 These genes include PAX5, E2A, EBF1, LEF1, IKAROS, and AIOLOS, and their mutations predominantly produce haploinsufficiency resulting in hypomorphic expression. Forty percent of acute precursor B-cell leukemias possessed mutation of at least one gene required for B-cell development, and 31.7% specifically had mutations within the PAX5 gene. Because of the requirement of these factors for normal early to late precursor B development, the immunophenotypic stage most closely related to the leukemias, it is hypothesized that loss of normal expression levels leads to a block in differentiation, a critical step in leukemogenesis.135
MUTATIONAL COMPLEMENTATION GROUPS IN ACUTE LEUKEMIAS
Several lines of evidence indicate that more than one mutation is necessary for the pathogenesis of acute leukemia. First, there is evidence for acquisition of additional cytogenetic abnormalities with disease progression from CML to AML (i.e., CML blast crisis). Published examples of progression in BCR/ABL-positive CML include acquisition of t(3;21) AML1/EV11, t(8;21) AML1/ETO, or t(7;11) NUP98/HOXA9 gene rearrangements. Progression of chronic myelomonocytic leukemia to AML in a patient with the TEL/PDGFβR gene rearrangement was associated with acquisition of a t(8;21) AML1/ETO gene rearrangement.136 Second, expression of the AML1/ETO or CBFβ/MYH11 fusion proteins in murine models is not sufficient to cause AML.27,137 Chemical mutagens must be used in these contexts to generate second mutations that cause the AML phenotype. Third, evidence indicates that in some cases the TEL/AML1 gene rearrangement associated with pediatric ALL may be acquired in utero, but ALL does not develop until years later, indicating a requirement for a second mutation.138 Fourth, AML develops in transgenic mice that express the PML/RAR-α fusion protein only after a long latency of 3 to 6 months, with incomplete penetrance, indicating a need for a second mutation.34–36
The genetic epidemiology of AML provides important clues to the nature of the collaborating mutations. One broad complementation group in AML (Fig. 31.1) is composed of mutations that activate signal transduction pathways. These include activating mutations in FLT3, RAS, and KIT and, more rarely, the BCR/ABL and TEL/PDGFβR fusion associated with disease progression in CML. These can be viewed as a complementation group because even though they are collectively present in approximately 50% of cases of AML, they rarely, if ever, occur together in the same patient.
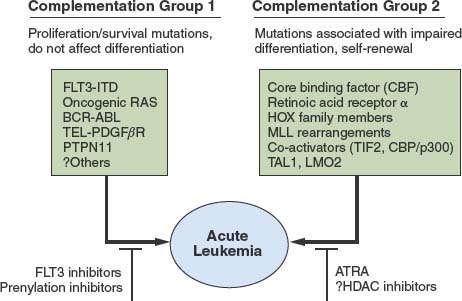
FIGURE 31.1 Cooperating mutations in acute leukemia. Leukemia is composed of two broad complementation groups, defined by lack of concurrence of any two mutations in the same complementation group in the same patient. One group is characterized by activating mutations in signal transduction pathways, such as FLT3-ITD or oncogenic N-RAS. When expressed alone, these mutations confer a proliferative or survival advantage, or both, but do not affect differentiation. The second group exemplified by AML1/ETO or PML/RAR-α are associated with impaired differentiation and the ability to confer properties of self-renewal to hematopoietic progenitors. Together, the complementation groups collaborate to engender the acute leukemia phenotype. This model has important therapeutic implications in that each of the complementation groups can be potentially targeted for therapeutic intervention, such as small-molecule inhibitors of FLT3 or RAS, or agents that override the block in differentiation, such as all-trans-retinoic acid (ATRA) or possibly histone deacetylase (HDAC) inhibitors.
A second complementation group, typified by translocations involving hematopoietic transcription factors, includes AML1/ETO, CBFβ/SMMHC, PML/RARα, NUP98/HOXA9, and MLL gene rearrangements, and MOZ/TIF2, and they are never observed together in the same leukemia. In general, this second class of mutations impairs hematopoietic differentiation and may confer properties of self-renewal to the leukemic stem cell but is not sufficient to cause leukemia when expressed alone (see “Recurring Chromosomal Abnormalities in Acute Leukemia”). However, one mutation from each of these two complementation groups often coexists in the same leukemia. For example, activating mutations in FLT3 or RAS have been observed in association with virtually all of the fusion genes in the second class described earlier.139
Support has also been given for the hypothesis of collaborating classes of leukemia oncogenes derived from analysis of genotypes of CML patients who progress to AML. Some cases of BCR/ABL-positive CML progress to AML associated with acquisition of the t(7;11) translocation associated with expression of the NUP98/HOXA9 fusion gene discussed earlier. As another example, TEL/PDGFβR-positive chronic myelomonocytic leukemia may progress to AML associated with acquisition of the t(8;21) translocation related to expression of the AML1/ETO fusion. These cases of disease progressions from CML to AML imply that constitutively activated tyrosine kinases cooperate with mutations in hematopoietic transcription factors to cause the AML phenotype.
These findings suggest a hypothesis for the pathogenesis of AML in which there are two broad classes of cooperating mutations (Fig. 31.1).139 One class, exemplified by activating mutations in FLT3 or RAS, confer either a proliferative or survival advantage, or both, to hematopoietic progenitors but do not affect differentiation. These mutations do not confer self-renewal capacity as assessed in part by the ability to serially replate in culture or to serially transplant disease in murine models.140,141 A second class of mutations, exemplified by AML1/ETO, CBFβ/SMMHC, PML/RARα, NUP98/HOXA9, and MLL gene rearrangements, and MOZ/TIF2 serve primarily to impair hematopoietic differentiation and confer properties of self-renewal. Together, these cooperating mutations induce the AML phenotype characterized by enhanced proliferative and survival advantage, impaired differentiation, and limitless self-renewal capacity. Experimental evidence supports this model of cooperativity in murine models between BCR/ABL and NUP98/HOXA9,142 TEL/PDGFβR and AML1/ETO,143 and FLT3/ITD and PML/RAR-α.144 These findings have important therapeutic implications in that it may be possible to target both classes of mutations. For example, in APL with activating mutations in FLT3, it may be possible to target FLT3 with small-molecule inhibitors and PML/RAR-α with ATRA (see Fig. 31.1).
CONCLUSION
The quest to elucidate the essential pathophysiologic changes involved in leukemogenesis has been accelerated with the use of newer technologies such as high-resolution mapping and high-throughput sequencing. It is now possible to identify specific molecular pathways complementing known recurrent translocations as well as gain insight into the mechanisms underlying normal karyotype leukemias. Not only can these novel mutations be used for more accurate prognostication, but they also provide an opportunity for drug development, targeting the essential pathways dysregulated in leukemia. As the availability of pathway-targeted therapeutics increases, interrogation of a patient’s leukemia for alterations at the genomic level may allow individualized therapy addressing the pathways responsible for leukemic cell survival, proliferation, and differentiation, which ideally would improve treatment efficacy and reduce therapy-related morbidity and mortality.
Selected References
The full list of references for this chapter appears in the online version.
1. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730.
3. Saito Y, Kitamura H, Hijikata A, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med 2007;2(17):17.
6. Huntly BJ, Shigematsu H, Deguchi K, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004;6:587–596.
7. Somervaille TCP, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006;10:257–268.
9. Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer 2005;5:311–321.
10. Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 2004;351:657–667.
13. Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008;456:66.
14. Loriaux MM, Levine RL, Tyner JW, et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood 2008;111:4788.
16. International Cancer Genome Consortium. International network of cancer genome projects. Nature 2010;464:993–998.
17. Rowley JD. The role of chromosome translocations in leukemogenesis. Semin Hematol 1999;36:59–72.
18. Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBP{alpha} differentiation pathway in human cancer. J Clin Oncol 2009;27:619–628.
26. Jiao B, Wu CF, Liang Y, et al. AML1-ETO9a is correlated with C-KIT overexpression/mutations and indicates poor disease outcome in t(8;21) acute myeloid leukemia-M2. Leukemia 2009;23:1598.
32. Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell 2002;108:165–170.
33. Scaglioni PP, Pandolfi PP. The theory of APL revisited. Curr Top Microbiol Immunol 2007;313:85–100.
37. Tallman MS, Nabhan C, Feusner JH, Rowe JM. Acute promyelocytic leukemia: evolving therapeutic strategies. Blood 2002;99:759–767.
38. Abramovich C, Humphries RK. Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol 2005;12:210–216.
45. Eguchi M, Eguchi-Ishimae M, Greaves M. Molecular pathogenesis of MLL-associated leukemias. Int J Hematol 2005;82:9–20.
46. Slany RK. The molecular biology of mixed lineage leukemia. Haematologica 2009;94:984–993.
51. Wei J, Wunderlich M, Fox C, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell 2008;13:483.
56. Kung AL, Rebel VI, Bronson RT, et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev 2000;14:272–277.
62. Mercher T, Raffel GD, Moore SA, et al. The OTT-MAL fusion oncogene activates RBPJ-mediated transcription and induces acute megakaryoblastic leukemia in a knockin mouse model. J Clin Invest 2009;119:852–864.
64. Sweetser DA, Chen CS, Blomberg AA, et al. Loss of heterozygosity in childhood de novo acute myelogenous leukemia. Blood 2001;98:1188.
65. Ebert BL, Pretz J, Bosco J, et al. Identification of RPS14 as a 5q(-) syndrome gene by RNA interference screen. Nature 2008;451:335.
69. Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet 1999;23:166–175.
71. Armstrong SA, Look AT. Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol 2005;23:6306–6315.
72. O’Neil J, Look AT. Mechanisms of transcription factor deregulation in lymphoid cell transformation. Oncogene 2007;26:6838–6849.
73. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009;458:719–724.
75. Lancet JE, Karp JE. Farnesyltransferase inhibitors in hematologic malignancies: new horizons in therapy. Blood 2003;102:3880–3889.
76. Braun BS, Shannon K. Targeting Ras in myeloid leukemias. Clin Cancer Res 2008;14:2249–2252.
84. Frohling S, Scholl C, Levine RL, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell 2007;12:501–513.
85. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006;108:3472–3476.
86. Hussein K, Bock O, Theophile K, et al. MPLW515L mutation in acute megakaryoblastic leukaemia. Leukemia 2009;23:852–855.
88. Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene 2007;26:6724–6737.
91. Walters DK, Mercher T, Gu TL, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell 2006;10:65–75.
92. Mullighan CG, Downing JR. Genome-wide profiling of genetic alterations in acute lymphoblastic leukemia: recent insights and future directions. Leukemia 2009;23:1209.
94. Hohenstein P, Hastie ND. The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet 2006;15:R196.
96. Yang L, Han Y, Saurez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia 2007;21: 868–876.
97. Miyagi T, Ahuja H, Kubota T, et al. Expression of the candidate Wilm’s tumor gene, WT1, in human leukemia cells. Leukemia 1993;7:970–977.
101. Summers K, Stevens J, Kakkas I, et al. Wilms’ tumour 1 mutations are associated with FLT3-ITD and failure of standard induction chemotherapy in patients with normal karyotype AML. Leukemia 2007;21:550–551.
102. Virappane P, Gale R, Hills R, et al. Mutation of the Wilms’ tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol 2008;26:5429–5435.
103. Paschka P, Marcucci G, Ruppert AS, et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B Study. J Clin Oncol 2008;26:4595–4602.
105. Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer 2002;2:594.
109. Venkatachalam S, Shi Y-P, Jones SN, et al. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J 1998;17:4657–4667.
110. Haferlach C, Dicker F, Herholz H, et al. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008; 22:1539–1541.
115. Seifert H, Mohr B, Thiede C, et al. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia 2009;23:656.
122. Radtke F, Wilson A, MacDonald HR. Notch signaling in hematopoiesis and lymphopoiesis: lessons from Drosophila. Bioessays 2005;27:1117–1128.
123. Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004;306:269–271.
124. Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer 2006;6:347–359.
125. DeAngelo DJ, Stone JR, Silverman LB, et al. A phase I clinical trial of the Notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. ASCO Meeting Abstracts 2006:6585.
126. Real PJ, Tosello V, Palomero T, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med 2009;15:50–58.
127. Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 2005; 352:254.
131. Mueller BU, Pabst T. C/EBPalpha and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol 2006;13: 7–14.
133. Wechsler J, Greene M, McDevitt MA, et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 2002;32:148–152.
134. Malinge S, Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood 2009;113:2619–2628.
135. Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007;446:758–764.
144. Kelly LM, Kutok JL, Williams IR, et al. PML/RARalpha and FLT3-ITD induce an APL-like disease in a mouse model. Proc Natl Acad Sci U S A 2002;99:8283.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


