I. EPIDEMIOLOGY AND ETIOLOGY
A. Incidence. Acute leukemia afflicts 3 to 4/100,000 population annually (11,000 new cases/year) in the United States. Children account for 25% of cases. Acute leukemia is the most common malignant disease of childhood.
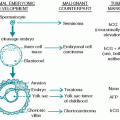
1. Cell type. Of cases of acute lymphoblastic leukemia (ALL), 80% occur in children, and 90% of cases of acute myelogenous leukemia (AML) occur in adults.
2. Age. In adults, AML rates begin to rise exponentially after 50 years of age; the age-specific incidence rate is 3.5/100,000 in adults 50 years of age, and increases significantly to 15 at age 70 and 35 at age 90. The mean age for AML in the United States is 63 years. A peak incidence of ALL occurs at 3 to 4 years of age; the incidence steadily decreases after 9 years of age and rises in incidence after 40 years of age. Despite being considered a childhood cancer, the age-related increase in incidence of most cancers also pertains to ALL.
3. Sex. Acute leukemia shows a male predilection only in very young and elderly patients.
B. Etiology
1. Hereditary
a. Hereditary syndromes that are associated with chromosomal abnormalities, a high risk of acute leukemia, and excessive chemosensitivity include the following:
(1) Bloom syndrome is a recessively transmitted disease occurring predominantly in people of Jewish ancestry. Chromosome breaks are readily found in cytogenetic studies. The syndrome is characterized by short and thin stature, delicate features, telangiectatic lesions over the malar eminences of the face, photosensitivity, and a variety of other cutaneous abnormalities (acanthosis nigricans, hypertrichosis, ichthyosis, and café au lait spots). AML is the type of leukemia that develops in these patients.
(2) Fanconi congenital pancytopenia (Fanconi anemia) is an autosomal recessive disease often associated with multiple chromosomal abnormalities. Clinical features include skeletal abnormalities (absence of radii, hypoplasia of the thumbs), squinting, microcephaly, small stature, and hypogonadism. AML, as well as skin carcinoma, often complicates this syndrome.
(3) Down syndrome (mongolism, trisomy 21) is associated with an increased risk for both AML and ALL.
(4) Ataxia telangiectasia is associated with an increased incidence of lymphoid malignancies, including ALL.
b. Siblings of younger patients with acute leukemia have a fivefold increased risk of developing leukemia. There is about a 15% concordance if one member of a pair of monozygotic twins develops AML, although this may be due to placental metastasis.
2. Radiation is a well-documented leukemogenic factor in humans. Increased incidence of leukemia proportional to the cumulative radiation dose has been demonstrated in populations exposed to atomic bombs, in patients irradiated for ankylosing spondylitis, and in radiologists (before current protective precautions). Doses <100 cGy are not believed to be associated with the development of leukemia. The types of leukemia induced by radiation are ALL, AML, and chronic myelogenous leukemia (CML), but not chronic lymphocytic leukemia.
3. Viruses have not been shown to be etiologic for acute leukemias in humans, although an association is seen with Epstein-Barr virus with ALL-L3 (Burkitt leukemia). HTLV-1 is discussed in
Chapter 21, in “Non-Hodgkin Lymphoma.”
4. Chemicals. The ability of chemicals to produce acute leukemia and pancytopenia is likely related to their ability to mutate or ablate the bone marrow stem cells.
a. Benzene and toluene were identified as carcinogens associated with acute leukemia a century ago. Acute leukemia develops 1 to 5 years after exposure and is often preceded by bone marrow hypoplasia, dysplasia, and pancytopenia.
b. Drugs. Drug-induced acute leukemia is usually preceded by myelodysplasia. Alkylating agents and topoisomerase II inhibitors given for prolonged periods are associated with a markedly increased risk of AML when compared with the general age-matched population. Exposure to arsenicals has also been implicated as an increased risk factor for leukemia development. Secondary AML currently accounts for 10% to 20% of all AML cases.
5. Hematologic diseases. Transformation into acute leukemia (“blast crisis”) is seen in >80% of cases of CML and is part of its natural history. Patients with myelodysplastic syndromes (MDS) clearly have an increased likelihood of evolution to AML. The incidence of AML in myeloproliferative disorders (MPD), myeloma, and certain solid tumors is increased by the use of chemotherapy.
6. Smoking. Cigarette smoking is associated with approximately 50% increase in leukemia risk. Cigarette smoking has a deleterious effect on survival in AML by shortening complete remission duration and subsequent survival. It has been associated with severe infections during aplasia. Leukemogenic compounds favoring complex karyotypic abnormalities could also be involved.
II. PATHOLOGY, CLASSIFICATION, AND NATURAL HISTORY OF ACUTE LEUKEMIA
A. Classification
1. Morphologic features of acute leukemias
a. The French—American—British (FAB) histopathologic classification of acute leukemia was originally proposed in 1976. This system has been supplanted by the World Health Organization (WHO) classification below. The FAB defined the M1—M7 and L1—L3 subtypes of acute leukemia as follows but is frequently left out in modern interpretations of AML:
b. Auer rods are abnormal condensations of cytoplasmic granules. Their presence in the immature cells distinguishes AML from ALL; their absence is not diagnostically helpful.
c. Cytologic features of the acute leukemia subtypes, particularly the nuclear configurations, cytoplasm granularity, and prevalence of Auer rods, are shown in
Appendix C7.
d. Cytochemistry. Identifying the type of early cell may be difficult, but it is facilitated by flow cytometry. Traditionally used histochemical techniques, particularly for myeloperoxidase and nonspecific esterase (see
Appendix C7), are seen in AML. Myeloperoxidase activity can be assessed by both cytochemistry and flow cytometry.
e. Immunologic markers assessed by flow cytometry usually distinguish ALL from AML as well as identify their subtypes. These markers are also summarized in
Appendix C7. Antibodies against platelet glycoproteins (CD41 or CD61) are useful in distinguishing megakaryocytic (M7) leukemia. Flow cytometry has largely replaced cytochemistry for classification of acute leukemias in most centers. Flow cytometry is most useful when using antibodies against panmyeloid antigens (CD13 and CD33), monocyte antigens (especially CD11b and CD14), and hematopoietic progenitor cell antigens (CD34 and HLA-DR).
2. The WHO classification has replaced the FAB classification. The FAB classification, which provided a consistent morphologic and cytochemical framework, did not reflect the cytogenetic or clinical diversity of the disease. The WHO classification system takes into account the developing knowledge of the biology of AML, its distinct subtypes divided into diseases characterized by proliferative biology and diseases characterized by disorders of maturation. The WHO classification of AML is as follows (with corresponding FAB designations and approximate proportions of AML cases in brackets):
a. AML with recurrent cytogenetic abnormalities (
Table 25.1)
AML with abnormal bone marrow eosinophils and inv(16)(p13;q22) or t(16;16)(p13;q22) [M4 Eo, 10% to 12%]
Acute promyelocytic leukemia and variants; t(15;17)(q21;q11) and its variants [M3, M3v; 5% to 8%]
AML with t(8;21)(q22;q22); (AML1/ETO) [M2, 5% to 12%]
AML with 11q23 (MLL) abnormalities [M5 or M1, 5% to 6%]
b. AML with multilineage dysplasia [M2 or M6]
Following MDS or MDS/MPD
Without antecedent MDS
c. AML and MDS, therapy-related (alkylating agents, topoisomerase inhibitors, other types)
d. AML not otherwise categorized AML minimally differentiated [M0, 5%]
AML without maturation [M1, 10%]
AML with maturation [M2, 30% to 45%]
Acute myelomonocytic leukemia [M4, 15% to 25%]
Acute monoblastic and monocytic leukemia [M5a, M5b; 5% to 8%]
Acute erythroid leukemia [M6, 5% to 6%]
Acute megakaryoblastic leukemia [M7, 3% to 5%]
Acute panmyelosis with myelofibrosis [M7, rare; “acute myelofibrosis”]
Acute basophilic leukemia (very rare)
e. Myeloid sarcoma (“chloroma,” “granulocytic sarcoma”; extramedullary masses of monoblasts or myeloblasts)
f. Acute lymphocytic leukemias are now included in the WHO classification of lymphoid tissues as “precursor B-cell lymphoblastic leukemia/lymphoma” and “precursor T-cell lymphoblastic leukemia/lymphoma” (see
Appendix C6). These were designated as L1, L2 or L3 subtypes of ALL in the FAB system.
3. The two most significant differences between the FAB and the WHO classifications are
a. A lower blast threshold for the diagnosis of AML: The WHO defines AML when the blast percentage reaches 20% in the bone marrow (rather than 30% as defined by FAB).
b. Patients with recurring clonal cytogenetic abnormalities should be considered to have AML regardless of the blast percentage: t(8;21)(q22;q22), t(16;16)(p13;q22), inv(16)(p13;q22), or t(15;17) (q22;q12).
4. A more clinically relevant classification of AML can be achieved if two distinct subgroups with different biologic features are recognized.
a. AML that evolves from MDS is associated with multilineage dysplasia, poor-risk cytogenetics, and a poor response to therapy. The incidence of this type increases with age and is consistent with the hypothesis that MDS and MDS-related leukemia results through multiple insults to the molecular constitution of the hematopoietic stem cell.
b. AML that arises de novo usually lacks significant multilineage dysplasia. It is often associated with favorable-risk cytogenetic findings and has a better response to therapy with a higher likelihood of failure-free and overall survival. This type of leukemia has a relatively constant incidence throughout life and is the type most likely to be observed in children and young adults. Some types are clearly related to disorders of differentiation such as AML characterized by core-binding-factor abnormalities, t(8;21) or inv(16) or t(15;17). Others are characterized more by proliferative defects such as AML with normal cytogenetics (often with FLT3 mutations).
B. Pathology. Bone marrow examination in acute leukemia demonstrates hypercellularity with a monotonous infiltration of immature cells. Normal marrow elements are markedly decreased. Erythroblast maturation is commonly megaloblastoid in all types of AML, especially subtype M6. Cytologic features of the AML subtypes are shown in
Appendix C7.
C. Natural history. Leukemia cells generally replicate more slowly than their normal counterparts. Hematopoiesis is disturbed even before the proportion of blast cells in the marrow is conspicuously increased. Immature and malfunctioning leukocyte progenitors progressively replace the normal bone marrow and infiltrate other tissues. Relapse is inevitable in most patients unless
complete remission after induction and consolidation therapy persists at least 4 years. Relapse is associated with progressively poorer response to therapy and, if second or subsequent remission is achieved, progressively shorter duration of remission. Unsuccessful therapy is usually followed by death within 2 months. Death in acute leukemia is usually caused by either infection or hemorrhage.
D. Biology of acute promyelocytic leukemia (APL)
1. Morphology. Classified as M3 in the FAB classification, APL is characterized morphologically by the presence of blasts cells with heavy azurophilic granules, bundles of Auer rods, and a bilobed or reniform nucleus. Although most APL cases fit the description of hypergranular blasts, a cytologic microgranular variant (M3v) has been identified. The blasts in M3v have a bilobed, multilobed, or reniform nucleus and, under the usual staining, are devoid of granules or contain only a few fine azurophilic granules. The apparent paucity of granules is a result of their submicroscopic size. M3v is commonly associated with hyperleukocytosis and accounts for 15% to 20% of APL cases.
2. Immunophenotyping. APL blasts are positive for CD33 and CD13 but negative for HLA-DR and usually have a low-level expression of CD34. M3v blasts tend to be positive for CD34, CD2, and CD19.
3. Cytogenetics. Both the classic and the M3v forms of APL are associated with a specific cytogenetic abnormality, t(15;17)(q22;q21). This translocation disrupts the PML gene on chromosome 15 and the retinoic acid receptor α (RARα) on chromosome 17, resulting in a fusion gene (PML/RARα). The protein product of PML/RARα retains the retinoic acid (RA) ligand-binding domain and plays a key role in leukemogenesis, as well as in mediating the response to retinoids. Expression profiling by microarray is likely to be used in the future as an adjunct to cytogenetics or as a replacement.
a. Four other alternative translocations associated with APL have been characterized:
t(11;17)(q23;q21) involving the PLZF gene on chromosome 11
t(5;17)(q35;q21) involving the NPM gene on chromosome 5
t(11;17)(q13;q21) involving the NuMA gene on chromosome 11
t(1;17) occurs but is rare.
b. PML/RARα-mediated APL is sensitive to retinoids, as are the variants NPM/RARα- and NuMA/RARα-mediated APL. In contrast, PLZF/ RARα-associated APL is considered resistant to retinoids as well as to arsenic trioxide.