Acquired Disorders of Coagulation: The Immune Coagulopathies
Charles R.M. Hay
The formation of a blood clot is the final result of a cascade of interactions among multiple plasma proteins that ultimately result in the conversion of fibrinogen to fibrin and the cross-inking of fibrin by activated factor XIII to form a stable clot. Quantitative or qualitative deficiencies of many of the coagulation factors involved in these reactions may be associated with a clinically significant bleeding disorder. Circulating immunoglobulin anticoagulants may arise in the plasma of individuals whose coagulation mechanism was previously normal. These inhibitor antibodies, usually but not exclusively immunoglobulin G (IgG), may function as a circulating anticoagulant when directed against a specific clotting factor protein. These are autoantibody inhibitors, in contrast to the alloantibody inhibitors, which arise in individuals with congenital factor deficiencies as a complication of replacement therapy.1,2
Although neutralizing autoantibody inhibitors have been described to neutralize each of the coagulation factor proteins, antibodies to factor VIII, von Willebrand factor (vWF), and factor X are rare, and autoantibodies to other clotting proteins are extremely rare. Nevertheless, when they do occur, they may produce life- and/or limb-threatening bleeding. Acquired hemophilia A (factor VIII:C deficiency) is the most common of the acquired autoantibodies against specific clotting factors but is much less prevalent than the lupus anticoagulant (LAC). In fact, the acquired autoantibody, which characterizes the LAC, is an in vitro anticoagulant only, and is actually associated with clinical hypercoagulability in vivo.
Autoantibodies targeting specific coagulation factor proteins are often associated with more severe bleeding manifestations and often demonstrate more complex pharmacokinetics and binding affinities than alloantibodies directed against the same protein. Although frequently found in subjects without any associated pathology, autoantibody inhibitors may arise in association with a number of benign diseases, such as autoimmune disorder, chronic inflammatory and infectious states, and in pregnancy. The development of autoantibody inhibitors may also complicate lymphoproliferative and, to a lesser degree, solid tumor malignancies. Incidental associations have also occurred in the context of certain antibiotics and bone marrow transplantation. Some associations are quite specific, for example, the association between myeloproliferative disease and hyperthyroidism and mild acquired von Willebrand disease (vWD) and the association between amyloid and factor X deficiency, but many patients have no obvious underlying pathology.
From a laboratory perspective, the diagnosis of autoantibody inhibitors is based on the inability of normal plasma to correct prolonged clotting assays produced by patient plasma in mixing studies. The potency of the inhibitor and the target of the inhibitor determine the mode of treatment of acute bleeding episodes.
ACQUIRED INHIBITORS TO FVIII
Acquired Hemophilia
Autoantibody FVIII:C neutralizing inhibitors appear spontaneously in subjects with previously normal hemostatic function causing acquired hemophilia.1,2,3 This rare immune coagulopathy occurs with an incidence rate of 1.38 and 1.48 per 1 million per year in the only two studies of acquired hemophilia in which the patients are linked to a defined population.4,5 The latter figure is as close to a true incidence as one can currently get, since it is based on a 2-year, cross-sectional, UK study in which considerable effort was expended to record all cases diagnosed in every hospital in the United Kingdom during that time. Even so, many low-titer (<5 BU) inhibitors may remain undetected unless patients experience severe bruising or bleeding after surgery or trauma. Some cases of mild disease may be missed because of the vagaries of diagnostic accuracy and the tendency for these patients to present to generalists, surgeons, and physicians. Acquired hemophilia is being diagnosed more frequently now that there is increased awareness that this syndrome complicates a wide variety of autoimmune and malignant diseases. Also, the availability of improved and specific treatment options for acquired hemophilia has increased the urgency of accurately diagnosing this potentially severe and morbid condition.
Epidemiology and Clinical Presentation
Acquired hemophilia has an equal sex distribution overall, though younger patients are more frequently female.5 The condition may arise at any time in life but usually presents in the extremely elderly,4,5,6,7,8,9,10,11 the median age at presentation having been reported by Collins to be 78 years (range 2 to 98 years).4 Earlier series tend to report relatively younger patient groups, either because of recall, referral, and/or reporting bias6,7 or because of the focus of the report is on the treatment of bleeding.8,9 Presentation during childhood is exceptionally rare.5,9,10
In approximately 50% of individuals with auto antibodies to FVIII:C, there is no obvious underlying disease state (“idiopathic” inhibitors).4,5,6,11 Older patients with acquired hemophilia are much less likely to have an associated disease state than younger patients.5 Diseases associated with acquired hemophilia include autoimmune diseases, hematologic and nonhematologic malignancies, drug reactions, and pregnancy.4,5,6 These are described in greater detail below:
There is a strong inverse correlation between the age at presentation and the likelihood that the acquired hemophilia has arisen in relation to some underlying disease.5 Collins found an underlying diagnosis in all five patients under the age of 40 years, 55% of those between 40 and 59 years, 42% of those between 60 and 79 years but only 23% of those aged 80 or over (P < 0.01).
Immune Disorders
Acquired hemophilia has not only been described particularly with chronic, long-standing, severe, rheumatoid arthritis, but also with other autoimmune disorders/collagen vascular diseases, including systemic lupus erythematosus (SLE), Sjögren syndrome, and less frequently with temporal arteritis, multiple sclerosis, myasthenia gravis, autoimmune hypothyroidism and hyperthyroidism, ulcerative colitis, graft versus host disease, and following vaccinations.5,6,10,11,12,13,14,15 Autoimmune dermatologic diseases, including pemphigus and psoriasis have also been occasionally associated with acquired hemophilia. Individuals with autoimmune FVIII:C antibody inhibitors secondary to autoimmune disease usually have high-titer inhibitors (>5 BU) that do not usually resolve spontaneously and are commonly relatively resistant to treatment with corticosteroids.
Malignancy
Hematologic malignancies, usually but not exclusively lymphoproliferative malignancies, have also occasionally been associated with acquired hemophilia. These include chronic lymphocytic leukemia (CLL), non-Hodgkin lymphoma, multiple myeloma, Waldenstrom macroglobulinemia, myelofibrosis, and myelodysplastic syndromes.5,6,17
Solid tumor malignancies, including carcinoma of the prostate, breast, lung, stomach, pancreas, colon, head and neck, and renal cell5,6,18 have all been associated with acquired hemophilia. Inhibitor antibodies secondary to solid tumors17,18 tend to be of low-titer. There is no correlation between the titer of the autoantibody to FVIII:C and tumor size, aggressiveness, or likelihood of tumor response to chemotherapy or radiation. Acquired hemophilia complicating malignancy has a poor overall prognosis. Successful suppression of the autoantibody inhibitor occurs in only about 20% of these cases and may require tumor eradication.17,18 If the autoantibody FVIII:C inhibitor does remit, it may recur and relapse of the inhibitor is not a reliable marker of tumor recurrence. Acquired hemophilia occurs much more frequently in association with lymphoproliferative malignancies than with solid tumors. CLL, non-Hodgkin lymphoma, multiple myeloma, and Waldenstrom macroglobulinemia are the most frequent hematologic malignancies associated with the development of autoimmune FVIII:C inhibitors. Although autoantibody and alloantibody inhibitors to FVIII:C are usually IgG, IgA and IgM monoclonal antibodies have been described in cases of multiple myeloma and CLL. Isolated cases of acquired hemophilia complicating myelodysplastic syndromes, myelofibrosis, and erythroleukemia, have also been reported.
Although acquired hemophilia has often been reported to follow drug administration, many of these drug associations may be coincidental and spurious.5 Nevertheless, the literature lists associations with allergic diseases and drug reactions including asthma; allergies to penicillin, sulfonamides, phenytoin, chloramphenicol, and methyldopa; treatment with fludarabine, interferon-α; and antipsychotic medications.19,20 Allergic reactions to medications, particularly sulfonamides, rarely penicillin and most recently clopidogrel,29 have been implicated in the development of an autoimmune response to FVIII:C. The inhibitory antibody usually disappears shortly after withdrawal of the offending drug. Of interest is the fact that acquired inhibitors to FVIII:C have also been observed after treatment with medications known to interfere with the immune system, such as interferon-α and fludarabine.19
Pregnancy
The association between acquired hemophilia and pregnancy has been overstated in earlier publications in which pregnancy-associated acquired hemophilia accounted for up to 10% to 15% of total cases. This is probably accounted for by recall and reporting bias. The recent 2-year survey of Collins et al.5 reported only three cases of pregnancy-associated acquired hemophilia (2% of the entire cohort and 4.3% of women reported). This represents 1 case per 350,000 births during that period. A similar incidence was reported from the Italian Registry.23
Acquired hemophilia arises in mostly primiparous women (80%), usually within 3 months postpartum, though cases have been described as late as 11 months postpartum, and presentation around the time of delivery or even antenatally is not uncommon.5,8,9,22,24,26 Of the 42 cases of pregnancy-associated acquired hemophilia reported to the European acquired haemophilia (EACH2) registry, only 1 presented antenatally.25 Late presentation may represent delayed diagnosis rather than late onset. Recurrence of the inhibitor in subsequent pregnancies has been reported but is not common, though mothers should be warned of this possibility. Although one study reported recurrence in four-sixth of subsequent pregnancies,21 no relapses were reported in nine subsequent pregnancies in a further report26 and no relapses were reported in the four patients reported in the Italian registry.23 When acquired hemophilia occurs antenatally, transplacental transfer of IgG autoantibody may cause life-threatening bleeding in the baby.23,27,28 Although spontaneous remission has been described in postpartum-acquired hemophilia patients, they usually require immunosuppressive therapy to achieve remission (see below). Severe uterine bleeding during labor or delivery may occur in those presenting antenatally and this sometimes requires hysterectomy. Nevertheless, the overall mortality of acquired hemophilia associated with pregnancy is relatively low (<5%).22,26 These cases are usually sensitive to corticosteroids alone, though spontaneous remission may occur in up to 76%, if steroids are withheld. This contrasts with the relatively low spontaneous remission rate of <30% observed generally for autoimmune hemophilia.22,23,24
Spontaneous remissions of the autoimmune antibody inhibitor targeting FVIII:C is not uncommon, and may occur in approximately one-third of cases, usually after months to years of involvement.6 More recent reports do not report spontaneous remission, because virtually all patients are offered immunosuppressive therapy to minimize the period during which they may be at hemorrhagic risk. Spontaneous remission in pregnancy-associated acquired hemophilia syndrome is said to be particularly common but, again, these women are usually offered immunosuppressive therapy to minimize the hemorrhagic risk.
Clinical Manifestations
The occurrence of an acquired inhibitor to FVIII:C typically is heralded by spontaneous, bruising and soft tissue bleeding in individuals who lack a previous bleeding history. Bleeding
may sometimes be life- and limb-threatening. Hemarthrosis are unusual in acquired hemophilia, in contrast to congenital hemophilia. The commonest bleeding manifestation of acquired hemophilia is bruising, with often massive ecchymosis. Muscle bleeds occur in 40%, followed in frequency by gastrointestinal, urinogenital, and retroperitoneal bleeding. Spontaneous bleeding into the central nervous system is fortunately relatively infrequent.5,6,7,8,9 Since the 1st obvious bleeding manifestation may be hematuria or melena, these patients may present to generalists and diagnosis may consequently be delayed, sometimes until after some surgical intervention.
may sometimes be life- and limb-threatening. Hemarthrosis are unusual in acquired hemophilia, in contrast to congenital hemophilia. The commonest bleeding manifestation of acquired hemophilia is bruising, with often massive ecchymosis. Muscle bleeds occur in 40%, followed in frequency by gastrointestinal, urinogenital, and retroperitoneal bleeding. Spontaneous bleeding into the central nervous system is fortunately relatively infrequent.5,6,7,8,9 Since the 1st obvious bleeding manifestation may be hematuria or melena, these patients may present to generalists and diagnosis may consequently be delayed, sometimes until after some surgical intervention.
Between 7.9% and 22% of cases have been reported to die from hemorrhage.6,7,8,25 More recent studies reveal a trend toward decreasing hemorrhagic mortality, which may be related to the availability of improved management options.8,9 However, the recent study of Collins et al. reported a 9.1% mortality rate from hemorrhage. Most hemorrhagic deaths are from gastrointestinal or retroperitoneal bleeding and occur within the 1st few weeks after presentation though patients remain at risk of life-threatening bleeding until the inhibitor has been eradicated.5 Delayed diagnosis and inadvertent treatment, such as surgery, contribute significantly to the early high mortality rate, occurring mainly because these patients frequently present to surgeons and general physicians.5,8
Infectious complications, secondary to immunosuppressive therapy, account for most of the late deaths in patients with acquired hemophilia. Indeed, infection appears to be a commoner cause of death than hemorrhage in acquired hemophilia. Collins reported an 11% mortality rate from sepsis.5 Indeed, 33% of patients from this cohort suffered from sepsis at some time in their course.
Although bleeding may be severe and life-threatening, the recent cross-sectional study of Collins showed that 34% of patients have only mild bleeding manifestations that did not require hemostatic replacement therapy.4,5 This 2-year study attempted to capture all cases of acquired hemophilia presenting in any hospital in the United Kingdom during a 2-year period. This observation also accounts for the paradoxical observation that the prognosis for acquired hemophilia is better for patients managed outside a major teaching center because those with severe bleeding are more likely to be referred in to a teaching hospital.5 Earlier reports tend to overstate bleeding severity either because of recall bias6 or because they only report patients treated for bleeding.8,9 Interestingly, at the time of presentation, neither the factor VIII level nor the inhibitor titer predicts the severity of subsequent bleeding events.5,8 Patients who died from hemorrhage and those who required no hemostatic therapy at all had a similar presenting median (range) factor VIII level and inhibitor titer.5
Laboratory Diagnosis of Autoimmune FVIII:C Antibody Inhibitors
The clinical suspicion of acquired hemophilia, based on the recent onset of significant bruising or bleeding in a previously noncoagulopathic individual, is strengthened by the detection of a normal prothrombin time (PT) but prolonged activated-partial-thromboplastin time (aPTT). The aPTT initially corrects using 50:50 normal plasma, which suggests a factor deficiency, but the aPTT subsequently prolongs again on incubation at 37°C. An intrinsic factor screen will reveal a low factor VIII level, typically lower than 0.05 IU/dL, but may appear to show less dramatic reductions in factors XI, XI, and XII. This is a laboratory artifact caused by the inhibitor-depleting factor VIII from the XI-, XI-, and XII-deficient plasma used in these one-stage assays. Dilution experiments show an apparent increase in the concentration of factors IX, XI, and XII, whilst the factor VIII level remains low.3,8,31,32 This may cause confusion with the LAC, which interferes nonspecifically with phospholipiddependant one-stage assays but whose clinical presentation is dramatically different.31 The diagnosis is confirmed using a formal Bethesda assay, possibly modified following the Nijmegen recommendations for low-titer inhibitors. Autoantibody factor VIII inhibitors in acquired hemophilia are usually polyclonal and have complex reaction kinetics. Consequently, patients with acquired hemophilia often have residual circulating factor VIII and the Bethesda assay is semiquantitative at best. This is linked with the clinical observation that bleeding severity is unrelated to the factor VIII level and inhibitor titer measured at the time of presentation. It is common practice to report the inhibitor titer based on the dilution closest to 50% inhibition. There have also been reports of anticoagulant treatment masking acquired hemophilia, possibly delaying diagnosis or being causally associated with the occurrence of acquired hemophilia. Patients on anticoagulants who present with bruising should therefore have both an aPTT and an International Normalized Ratio measured, for that reason.33,34,35 An enzyme-linked immunosorbent assay (ELISA) assay may be useful in complicated cases.3,30
Immunochemistry of Autoantibody Inhibitors to FVIII:C
In contrast with the oligoclonal alloantibodies found in congenital hemophilia, and which are usually both IgG1 and IgG4, autoantibody inhibitors to FVIII:C is usually polyclonal IgG4. A further distinctive feature is that the autoimmune autoantibody inhibitors may rarely be monoclonal IgA or IgM antibodies and shifting immunoglobulin profiles have been observed, for example, shifting from IgM to IgG.36
Systematic epitope mapping has indicated that autoantibodies are usually directed against either the A2 or C2 domain of the FVIII:C molecule (62%) and much less frequently to both epitopes as observed with alloantibodies, which overwhelmingly interact with both A2 and C2 domains.36,37 There appears to be preferential targeting of FVIII:C autoantibodies to the C2 domain (67%). The A3 epitope is targeted much less commonly. Both alloantibodies and autoantibodies bind to the same epitopes on the A2 and C2 domains. Autoantibodies may exert their inhibitory activity by interfering with thrombin cleavage sites on A2 or by impeding the interactions between FVIII:C and phospholipid or vWF protein on C2. This may result in increased FVIII:C catabolism and increased susceptibility of FVIII:C to degradation by activated protein C and factors IX and X.
Clinical Management
The management of acquired hemophilia involves treatment of any bleeding complications, simultaneous immunosuppression to abolish the inhibitor and investigation for and treatment of any underlying causative disorder. Patients with acquired hemophilia should be managed in or at least in collaboration with a hemophilia center experienced in managing the disorder, even if the initial presentation appears relatively benign. Invasive procedures and surgery should be avoided, if possible. Intramuscular injections should be avoided and venepuncture minimized since these can cause significant and troublesome
bleeding. If procedures or surgery are unavoidable, they should be conducted in a hospital with an experienced hemophilia center and with adequate hemostatic cover. Patient education is also important, especially for patients managed as an outpatient. The patient and their relatives have to be made aware of the seriousness of the condition so that they present to hospital in a timely manner.3,38,39
bleeding. If procedures or surgery are unavoidable, they should be conducted in a hospital with an experienced hemophilia center and with adequate hemostatic cover. Patient education is also important, especially for patients managed as an outpatient. The patient and their relatives have to be made aware of the seriousness of the condition so that they present to hospital in a timely manner.3,38,39
Management of Bleeding Complications
FIGURE 57.1 shows an algorithm for treatment of bleeding complications.
Bleeding may be life- and limb-threatening. Morbidity and mortality are reduced by prompt and adequate hemostatic treatment supervised at an experienced hemophilia center. None of the hemostatic options available have predictable efficacy in acquired hemophilia. Neither the presenting inhibitor titer nor the residual factor VIII level, nor the factor VIII level achieved following the infusion of human or porcine factor VIII are predictive of clinical response.3,8,9 Regular clinical review and appropriate imaging and laboratory monitoring are therefore vital when monitoring the efficacy of hemostatic therapy for this condition. An early assessment of hemostatic efficacy is very important so that if the treatment is not working a different agent may be tried with the minimum of delay. Dosing needs to be adequate and should continue for some time after bleeding is controlled, at a reducing dose, to optimize clinical response and minimize the risk of recurrent bleeding. This is particularly important for intracranial, intramuscular, and retroperitoneal bleeding. In contrast, many patients need no hemostatic treatment at all and subcutaneous bleeding and bruising can be managed conservatively, even if extensive. Mucosal bleeds may also benefit from the use of antifibrinolytic agents such as tranexamic acid or aminocaproic acid.
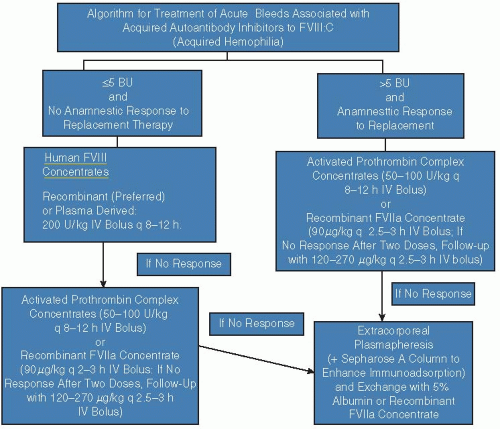 FIGURE 57.1 Algorithm for treatment of acute bleeds associated with acquired autoantibody inhibitors to FVIII:C. FIX, factor IX; BU, Bethesda units; rFVIIa, recombinant factor VIIa; PCC, prothrombin complex concentrate; DDAVP, 1-deamino-8-D-arginine vasopressin. |
Bleeding is treated either with bypassing agents (currently, Factor Eight Inhibitor Bypassing Activity [FEIBA] (Baxter, Vienna) or NOVOSEVEN (rVIIa, Novo, Denmark) or with factor VIII-containing agents to raise the factor VIII level.3 A recent survey from the EACH2 registry suggests that bypassing agents may be clinically more effective than factor VIII.40,41 Porcine factor VIII has been unavailable for some years, however, and this comparison may have to be reevaluated when the current recombinant B-domain-deleted porcine factor VIII completes ongoing clinical trials in acquired hemophilia and gains a product license for this indication.
Factor VIII:C Concentrates
Previous reports of the treatment of bleeding in acquired hemophilia have shown that human factor VIII concentrate is the 1st agent used to treat bleeding in about one-third of cases.6,8,9 The EACH2 registry data suggest that the response to factor VIII may be significantly worse than for bypass therapy in this group, probably because the half-life of factor VIII is severely curtailed in acquired hemophilia.40,41 Human factor VIII may be used in large doses, when this is sufficient to overcome a relatively low-affinity inhibitor and an effective postinfusion factor VIII increment can be achieved. The use of factor VIII is, therefore largely limited to patients with low-affinity inhibitors. Although dose-calculating formula have been proposed, empirical dosing combined with close laboratory monitoring of the response and prompt dose adjustment is probably the best approach since the
Bethesda titer is not quantitative in this condition and therefore gives a poor guide to the likely response to factor VIII. Factor VIII replacement combined with immunoadsorption of the inhibitor antibody may also be effective, if this is available.42,43 Human plasma-derived FVIII:C concentrates, which are characterized as being of intermediate purity by virtue of their substantial content of vWF protein, theoretically may be less susceptible to inactivation by the autoimmune FVIII:C inhibitor directed against the C2 domain37; however, this premise has not been confirmed in clinical studies. In contrast to congenital hemophilia, the use of factor VIII concentrate will not cause an anamnestic increase in the inhibitor titer.
Bethesda titer is not quantitative in this condition and therefore gives a poor guide to the likely response to factor VIII. Factor VIII replacement combined with immunoadsorption of the inhibitor antibody may also be effective, if this is available.42,43 Human plasma-derived FVIII:C concentrates, which are characterized as being of intermediate purity by virtue of their substantial content of vWF protein, theoretically may be less susceptible to inactivation by the autoimmune FVIII:C inhibitor directed against the C2 domain37; however, this premise has not been confirmed in clinical studies. In contrast to congenital hemophilia, the use of factor VIII concentrate will not cause an anamnestic increase in the inhibitor titer.
Porcine Factor VIII
Another source of FVIII:C concentrate, which was very much more effective than human factor VIII, was porcine factor VIII concentrate. Cross-reactivity of antibodies in acquired hemophilia to porcine factor VIII was characteristically low, averaging 5% to 10%,8,44,45 permitting porcine factor VIII to achieve hemostatic factor VIII levels in patients completely resistant to human factor VIII. Cross-reactivity varies considerably between individuals however, and so it is recommended to give an initial bolus of 75 to 100 IU porcine FVIII:C with subsequent dosing and dose intervals determined by the level of FVIII:C activity achieved and the durability of the response. FVIII:C levels of at least 30% to 50% are desirable, though clinical response has not been shown to correlate with the factor VIII level achieved.8 Porcine factor VIII was reported to give a good or excellent response in 78% of 74 bleeds and failure in only 9%.8 This response rate is comparable with that achieved using bypassing agents. More consistent high levels and improved pharmacokinetics may be achievable by administration using continuous infusion.46 In contrast to congenital hemophilia, an anamnestic rise in the factor VIII inhibitor is not seen following exposure to factor VIII in acquired hemophilia. Specific antiporcine factor VIII antibodies may arise in 30% and these patients, however, rendering them resistant to porcine factor VIII.45 Porcine factor VIII was manufactured from porcine plasma, which could not be rendered “virus free.” Consequently, the product was withdrawn some years ago. An alternative, recombinant B-domain-deleted porcine factor VIII (Inspiration, California) has recently completed trials in congenital hemophilia47 and multinational trials have begun in acquired hemophilia. This product looks very promising for the management of bleeding in acquired hemophilia but will probably not be available for general clinical use until 2013 to 2014.
Desmopressin (1-deamino-8-D-Arginine Vasopressin, DDAVP)
Administration of 1-deamino-8-D-arginine vasopressin (DDAVP) (Desmopressin) either intravenously or subcutaneously at the dose of 0.3 µg/kg induces a rapid rise of vWF and FVIII:C activities in normal individuals and those with mild uncomplicated hemophilia A and vWD.47 DDAVP may also induce a rapid rise of FVIII:C in individuals with low-titer inhibitors, rendering the drug suitable for the treatment of minor, non-life-threatening bleeds or for prevention of potential bleeding secondary to mild surgical procedures in the context of acquired hemophilia.48 The response is very variable, however. Of the 22 treatments reported, 11 patients with FVIII <5 IU/dL DDAVP resulted in an increase to between 15 and 140 IU/dL. Five patients with a Bethesda titer <2 BU/mL all responded with peak FVIII levels >80 IU/dL and a half-life of 4 to 6 hours. In contrast, no significant clinical response was reported for seven patients with a FVIII level <1 dL/mL.48 The use of DDAVP in this condition is therefore very limited, and it should probably therefore be reserved for the treatment of minor bleeding in patients with a discernible circulating factor VIII level and low-level inhibitors.3
Bypassing Agents
At present the choice of bypassing agents is limited to FEIBA (Baxter Vienna) and Novoseven (recombinant activated factor VII, rVIIa, NovoNordisk, Denmark), although several manufacturers are currently developing analogs of rVIIa. Although nonactivated prothrombin complex concentrates were used from the 1970s, these products have now largely fallen out of use for bleeding in acquired hemophilia. Both FEIBA and Novoseven have been shown to be effective in treating bleeding in acquired hemophilia.9,40,41,49,50,51,52
Although there are no direct comparative trials of FEIBA and Novoseven in acquired hemophilia, the data from the EACH2 registry and comparison of the published literature suggests that the two agents have similar efficacy.9,40,41,49,50,51,52 National statistics suggest that FEIBA is used more frequently than Novoseven for acquired hemophilia, at least in the United Kingdom.53 The choice of bypassing agent should depend, where possible, on the patient”s previous response, the dosing schedule, and possibly cost. When first-line therapy fails, the alternative bypassing agent may be used. In cases of life- and limb-threatening bleeding, both bypassing agents may be used sequentially or together since they synergize and have greater hemostatic efficacy when used in combination.53,54 The use of combination bypass therapy may be associated with an increased risk of both venous and arterial thrombosis, particularly in the elderly, and should therefore only be used for very serious bleeding, unresponsive to full doses of one or both bypassing agents.3
Both agents have been associated with a relatively small thrombotic risk.39,52,56,57,58,59,60 Disseminated intravascular coagulation (DIC), myocardial infraction, and deep vein thrombosis have been associated with the use of FEIBA.56 It is impossible to compare the thrombogenicity of the two products directly because most of the thrombotic events reported in relation to Novoseven are thought to relate to off-label use in nonhemophiliacs and FEIBA is currently only used in acquired and congenital hemophilia. However, the overall risk of thrombosis is low (~1 per 104-5 infusions). Twelve (8.6%) of 139 patients with acquired hemophilia treated with rVIIa were reported to have had a thrombotic event, which was arterial in 10 cases.52 Whether these events were directly related to rVIIa treatment is unclear and the authors point out that the methodology used would tend to overestimate the incidence of events.52 EACH2 reported 8 (2.3%) arterial events amongst 337 patients.40 One might expect that the thrombotic risk would be greater in patients with acquired rather than congenital hemophilia since the former are far more elderly and may have multiple riskfactors for arterial disease.
A limitation of bypassing agents is that they have no validated and readily available laboratory monitoring technique and dosing and dosing intervals must be adjusted based on clinical evaluation alone. Thrombin generation assays60,61 and thromboelastography (TEG)62,63 have both been used in a research
setting to assess response and adjust dosage of bypassing agents. Neither is the test fully validated nor widely available and there are no published data in relation to the use of these tests to manage acquired hemophilia. It may not be possible to extrapolate evidence on the use of these tests from congenital to acquired hemophilia because of the differing immunochemistry and kinetics of the inhibitors involved. Further studies are required. Furthermore, preliminary evidence suggests that TEG does not predict clinical response consistently.64
setting to assess response and adjust dosage of bypassing agents. Neither is the test fully validated nor widely available and there are no published data in relation to the use of these tests to manage acquired hemophilia. It may not be possible to extrapolate evidence on the use of these tests from congenital to acquired hemophilia because of the differing immunochemistry and kinetics of the inhibitors involved. Further studies are required. Furthermore, preliminary evidence suggests that TEG does not predict clinical response consistently.64
FEIBA
The postulated mechanism of action of FEIBA is that of “bypassing” the obstruction to coagulation caused by the neutralizing inhibitor of FVIII:C. This is accomplished through their content of trace amounts of serine proteases (activated factors IX, X, VII, and thrombin). Retrospective analyses of patient cohorts with alloantibodies and autoantibodies have revealed efficacy rates of approximately 80% for both FEIBA (average dose of 70 IU/kg).49 Furthermore, bolus doses higher than 200 IU/kg or frequent repeat dosing of activated prothrombin complex concentrates (aPCC) significantly increases the risks of thrombotic adverse events.56 A more recent retrospective report of 34 severe and moderate bleeds treated mainly with 75 U/kg between 8 and 12 hourly reported an efficacy of almost 100% after a median of 6 infusions for moderate bleeding and 10 infusions for severe bleeding.50 The EACH2 registry reported 94% efficacy for 64 bleeds treated with FEIBA.40
Recombinant FVIIa Concentrate
Recombinant FVIIa (rFVIIa, Novoseven) is generally administered as an intravenous bolus, resulting in supraphysiologic levels of FVIIa activity in blood. In individuals with normal coagulation, it is hypothesized that thrombin is generated after FVIIa interacts with tissue factor (TF) on mononuclear cell surfaces (monocytes and macrophages) and subsequently activates FX to participate in the tenase and prothrombinase complexes. This small quantity of thrombin is adequate only to activate platelets and to cleave vWF protein from FVIII:C, but this results in a self-amplification process in which rFVIIa binds to the surface of the activated platelet, activates FX in the presence of FVIII and FIX, and produces an extraordinary “thrombin burst” to affect fibrin formation.65 Pertinent to the clinical situation with autoantibody FVIII:C or FIX inhibitors, large amounts of administered rFVIIa may bind nonspecifically to the platelet surface and activate FX in a TF-independent manner and in absence of FVIII:C and/or FIX.
Novoseven is normally given in a dose of 90 µg/kg, administered every 2 to 3 hours because of its short circulating half-life but a larger dose of 270 µg/kg has been shown to be safe and as effective as three 2-hourly doses of 90 µ/kg.66,67 Once bleeding has stopped, the dose interval is lengthened but replacement therapy with rFVIIa should continue for several days to minimize the risk of re-bleeding. In the largest clinical trial in acquired hemophilia, rFVIIa, employed as first-line replacement therapy, reversed symptomatic bleeding 100% of the time, whereas rFVIIa, used as salvage or rescue therapy, yielded a 75% response rate.25 A recent systematic review52 reported a 95% efficacy for rVIIa, when used as first-line therapy for 103 bleeds. The EACH2 registry reported similar 91% efficacy for 170 bleeds treated with Novoseven as first-line therapy.40 When used as second line therapy, the reported efficacy of Novoseven is lower at 86%.52 The median duration of treatment has been reported to be 6 days (1 to 33).52 If bleeding is not successfully stopped within the initial 24 hours of treatment with rFVIIa, the patient is likely to remain unresponsive to further rFVIIa and an alternative regimen should be considered.
Overall, very few side effects, including episodes of thrombosis, have been reported for rFVIIa concentrate use in autoimmune hemophilia. A finite risk does exist, however, and cases of myocardial infarction, DIC, and venous thromboembolism (VTE) have been reported.40 No laboratory methods are available to monitor the hemostatic efficacy or safety of rFVIIa.
Immunosuppression Regimens
As soon as the diagnosis of acquired hemophilia has been established, the patient should be given some form of immunosuppressive treatment to eradicate the inhibitor.3,30,38 Although up to 25% to 30% of patients may remit spontaneously,22,23,24,68 they may suffer significant morbidity whilst awaiting spontaneous remission and are more likely not to remit. For that reason, the current consensus of opinion is that all patients with acquired hemophilia should be offered immunosuppressive therapy in the hope that this will not only optimize the remission rate but reduce the time taken to achieve remission, thus minimizing bleeding morbidity and mortality. However, when choosing an immunosuppressive regime, it should be remembered that infection is actually the commonest cause of death in acquired hemophilia5 and aggressive immunosuppressive regimens may contribute to the morbidity and mortality associated with the condition. For this and other reasons, aggressive immunosuppressive regimens have fallen a little out of favor in recent years as a broad generalization. The options available for immunosuppression include administration of: corticosteroids alone; corticosteroids plus cyclophosphamide; rituximab (anti-CD20 monoclonal antibody); cyclosporin A; immunosuppression/immune tolerance induction regimens; high-dose intravenous immunoglobulin (IVIG); plasmapheresis/immunoadsorption; immune-tolerance regimens, and finally, corticosteroids combined with one or more cytotoxic agents cytotoxic agents (e.g., Cyclophosphamide, Azathioprine, Vincristine, 6-MP).
The various available regimens are difficult to compare directly because patient selection and end-points vary from report to report and most reports are of uncontrolled cohort studies. Almost all are case-reports, single center cohorts, or retrospective surveys. Furthermore, since most reports come from teaching centers, there is reporting bias toward more seriously affected cases. There may also be positive publication bias since failures are more difficult to publish.
Although there are no reports of relapse in older publications, patients with acquired hemophilia have presumably always relapsed and recent studies report a relapse rate of 10% to 20%.5,69 Relapse occurred after a median of 7.5 months (0.25 to 14 months) in 20% of 102 patients.5 This finding was reproduced in the EACH2 registry, which reported 18% relapse amongst those treated with steroids alone and 12% relapse in those treated with steroids and cyclophosphamide after a median of 4 months. All patients with acquired hemophilia therefore require long-term follow-up, even if they go into complete remission and should be warned of the possibility of recurrent bleeding symptoms. Furthermore, many patients require long-term maintenance immunosuppressive therapy to maintain remission or even partial remission and to prevent relapse.
Corticosteroids with/without Cytotoxic Agents
For many years it was thought that prednisolone combined with cyclophosphamide offered a slightly higher remission rate and more rapid response, though this has recently been challenged.5 Recent data and reexamination of the literature led one to the conclusion that the combination had no advantages over prednisolone alone, however.3,5,41 The only randomized comparison recruited only 31 patients and was therefore underpowered. The patients were treated with prednisolone 1 mg/kg/d for 3 weeks. The 21 nonresponders were then randomized either to continue prednisolone or to continue prednisolone combined with cyclophosphamide.70 There was no difference between treatment arms, though the number of subjects was very small.
The UK prospective cohort study5 permitted a prospective but nonrandomized comparison of patients treated with prednisolone or with prednisolone plus cytotoxic agents. The study design (an all-inclusive cohort study of all patients presenting in the United Kingdom over 24 months) should prevent selection bias. This showed that the 34 patients treated with prednisolone alone had a 76% complete remission rate after a median of 49 (31 to 62) days treatment, compared to a 78% complete remission rate after 39 (34 to 57) days for 45 patients treated with steroids and cytotoxics. Neither the remission rate nor the time to remission were significantly different, though the comparison is probably, again, statistically underpowered.
Aggregation of data from uncontrolled cohorts suggested similar results.41 However, a systematic review31 of 20 publications showed that there was a higher remission rate in patients treated with prednisolone combined with cyclophosphamide. This was not associated with a lower mortality rate, however, perhaps because of the greater toxicity of the combination than prednisolone alone.31 An analysis of the 331 patients reported to the EACH2 registry has been conducted, in which the groups were matched for potential confounding variables such as age, gender, inhibitor titer, factor VIII level, and underlying etiology.71 This group reported an odds ratio (95% CI) of 3.25 (1.51 to 6.96) P < 0.001 in favor of combination therapy. There were peculiarities in this data in that the remission rate for steroids alone was much lower, at 58%, than reported in the aggregate literature, whereas the 80% remission rate reported for combination therapy is comparable with what has been published before if not higher than average. This difference was not reflected in outcome. Mortality and sustained remission rate was similar in both groups.5,31,71 This suggests that although combination treatments offer a better short-term remission rate they do not improve long-term outcome.71 The median time to remission is reproducibly 5 weeks, so treatment must be given for more than 3 weeks before an assessment of success or failure may be made.
Rituximab
Rituximab (anti-CD20 antibody) has been a popular treatment for acquired hemophilia, particularly since the publication of a report of its use in three patients in combination with other drugs72 and the first-line use of rituximab in 10 patients, of whom eight achieved rapid complete remission.73 The remaining two patients responded to cyclophosphamide. A further study reported a complete remission rate of 100% in six patients treated with rituximab combined with prednisiolone with or without cytotoxics after response times of 1, 2, 4, 8,36, and 52 weeks, which are similar to those reported for other regimens.74 Rituximab has proved to be a disappointing drug used as a second-line agent and there is no convincing evidence that is superior to other regimens.31,75 Franchini38 reviewed 71 patients with acquired hemophilia treated with rituximab, mostly in combination with a variety of other immunosuppressive agents. Although these authors reported a response rate in excess of 90% the data was difficult to interpret with confidence and they still suggested that rituximab should be used as a second-line agent in combination with steroids. The EACH2 data would support such a cautious approach since only 30 of the 51 patients (59%) treated with rituximab in combination achieved a stable remission, no better than steroids alone.70 Only 42% of the patients treated with rituximab alone achieved remission.70 Since rituximab is usually given in combination with one or more other immunosuppressive drugs, the data is difficult to interpret. However, there is no convincing evidence to suggest that it improves the remission rate or reduces the relapse rate and is therefore currently not recommended as first-line therapy.
Cyclosporine
Intravenous High-Dose Immunoglobulin
High-dose immunoglobulin enjoyed a vogue during the late 1980s and early 1990s, when reports of its successful use 1st appeared.78 The early promise was never realized, however. A prospective study of 16 patients treated with IVIG reported that only three subjects (one also treated with steroids) achieved a remission and all three had inhibitor titers ≤1 BU/mL.79 This 12.5% response rate is actually lower than the reported spontaneous remission rate.68 Neither the UK Cohort study5 nor the EACH2 registry nor the systematic review of Delgado et al.31 show any additional benefit from including IVIG in the treatment regimen for acquired hemophilia and so high-dose immunoglobulin is currently not recommended for this condition.3
Extracorporeal Immunoadsorption
Forty-five patients were reported to have been treated with a combination of cyclophosphamide, prednisolone, IVIG, and daily immunoadsorption using a sepharose column combined with 200 IU/kg of factor VIII daily.80 Bleeding was controlled rapidly and the inhibitor titer also declined rapidly achieving undetectable levels after a median of only 3 (95% CI 2 to 4) days. Remission was reported in 88% of patients after a median of 14 days. This is clearly a very effective approach to the management of bleeding, which may be useful in the treatment of resistant bleeding in centers equipped for immunoadsorption. The response remission rate is not convincingly superior to conventional therapy, however, and the observations are uncontrolled. The rationale for the use of the factor VIII in this regimen, other than to control bleeding is unclear. Since patients with acquired hemophilia do not usually elicit an anamnestic increase in their inhibitor following factor VIII infusion, it is difficult to understand in what way factor VII will act as an immunomodulatory agent in such a regimen.
Immune Tolerance Induction
The rationale for immune tolerance induction is that factor VIII may stimulate antibody-producing cells to divide, thus making them susceptible to cytotoxic immunosuppressants.81 This
hypothesis seems implausible given that the patient is making factor VIII constantly anyway and given that an anamnestic increase in inhibitor titer does not usually follow factor VIII infusion in acquired hemophilia. None of the studies of this approach were controlled and factor VIII was invariably administered in combination with an aggressive combination of cytotoxic immunosuppressive drugs and so the results are very difficult to interpret. Lian et al. reported a 92% remission rate in 12 patients treated with steroids, vincristine, and cyclophosphamide given thrice weekly with factor VIII after 1 to 3 cycles of this treatment.79 This group subsequently reported very similar results (83% remission) in six patients treated with the same immunosuppressive regimen but without factor VIII.82
hypothesis seems implausible given that the patient is making factor VIII constantly anyway and given that an anamnestic increase in inhibitor titer does not usually follow factor VIII infusion in acquired hemophilia. None of the studies of this approach were controlled and factor VIII was invariably administered in combination with an aggressive combination of cytotoxic immunosuppressive drugs and so the results are very difficult to interpret. Lian et al. reported a 92% remission rate in 12 patients treated with steroids, vincristine, and cyclophosphamide given thrice weekly with factor VIII after 1 to 3 cycles of this treatment.79 This group subsequently reported very similar results (83% remission) in six patients treated with the same immunosuppressive regimen but without factor VIII.82
Nemes and Pitlik83 reported on the use of a more aggressive regimen where factor VIII was administered daily (30 IU/kg) in a reducing regimen over 3 weeks combined with cyclophosphamide and methyl prednisolone. Although they reported a 93% remission rate after a median of 4.6 weeks compared to a 67% remission at a median of 28.3 weeks in six historical controls, the control group is inadequate since the number of subjects and controls is too small to provide an adequate comparison. The results of these studies do not provide sufficient evidence of additional benefit from the inclusion of factor VIII in the immunosuppressive regimen to recommend this approach.
In conclusion, although there is general agreement that all patients with acquired hemophilia should be offered immunosuppressive therapy to attempt to eradicate the inhibitor, in the absence of clinical trials, there is no consensus as to the best regimen to use. Furthermore, the choice of regimen does not appear to influence the prognosis. Prednisolone combined with cyclophosphamide is probably superior to prednisolone alone. Rituximab and cyclosporine are probably second-line approaches and combined cytotoxic regimens are probably third-line. A balance must be struck, in this predominantly elderly population, between adequate immunosuppression on the one hand and excessive treatment-related morbidity on the other.
INHIBITORS OF VON WILLEBRAND FACTOR PROTEIN
Acquired von Willebrand Disease
Autoantibodies to vWF protein give rise to a clinical disorder resembling congenital vWD, acquired von Willebrand disease or syndrome (AvWS). In the absence of any large prospective cross-sectional studies, the incidence of this condition is unknown and it is almost certainly also still under-diagnosed.84,85 The condition may arise at any stage of life but occurs most frequently in the elderly at a median age of 62 years (range 2 to 96 years.86 AvWS is usually, but not invariably, an epiphenomenon of an underlying disease, most commonly lymphoproliferative or myeloproliferative disorders, which account for between 48% and 63% of cases.85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108 The condition is seen in a wide variety of lymphoproliferative disorders including lymphoma, monoclonal gammopathy of unknown significance (MGUS), myeloma, and Waldenstrom macroglobulinemia.86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103 Mohri et al.88 reported acquired hemophilia in 10% of 260 prospectively studied patients with hematologic disorders. AvWS has also been reported in association with autoimmune diseases such as SLE85,86,109,110,111,112 and hypothyroidism,86,113,114,115,116,117 cardiovascular disorders86,118,119,120,121,122,123,124,125,126,127 and solid tumors.85,86,128,129,130 The association with cardiovascular disorders, in particular, has been increasingly recognized, accounting for 46% of cases in one recent retrospective study.118 Since 1st being recognized in association with congenital heart disease,119,120 AvWS has also been described in relation to a variety of acquired cardiac defects including aortic stenosis with gastric angiodysplasia (Heyde syndrome) and ventricular assist devices.121,122,123,124,125,126,127
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


