ABNORMAL SECRETION OF GROWTH HORMONE
DEFICIENCY OF GROWTH HORMONE
GH deficiency results from various causes, and the clinical manifestations depend on the age of the patient when the disease first occurs. The condition may be hereditary or acquired, and GH deficiency may be isolated or combined with other pituitary hormone deficiencies. Subdividing GH deficiency into childhood onset and adult onset categories is useful, because some of the clinical manifestations are different.
ETIOLOGY
Congenital Growth Hormone Deficiency.
Table 12-3 lists the causes of GH deficiency. Among the congenital causes, between 5% and 30% have a familial pattern, which suggests a genetic basis. Known genetic causes of combined pituitary hormone deficiency involve the transcription factors Prop-1 and Pit-1, both members of the POU-homeodomain family of proteins critical for tissue differentiation. These genes are sequentially expressed during pituitary ontogeny, and Pit-1 is dependent on the expression of Prop-1 (of which the full name is “Prophet of Pit-1”). Inactivating mutations in the PROP1 gene lead to defective pituitary development, with TSH, luteinizing hormone, follicle-stimulating hormone, GH, and prolactin deficiency.32 A two-base deletion in a hot spot in the
PROP1 gene, with resulting frameshift, is the most common cause.33 The clinical phenotype is one of familial dwarfism, with hypothyroidism and hypogonadism. Of interest, pituitary enlargement frequently occurs in affected patients—a finding whose pathogenesis is not clear. Mutations in the PIT1 gene are another cause of combined pituitary hormone deficiency.34 At least eight different mutations have been described; most are recessively inherited, but some are dominant negative (i.e., the abnormal gene product interferes with that derived from the normal allele). Pit-1 is important for development of somatotropes, lactotropes, and thyrotropes; affected patients suffer from GH, prolactin, and TSH deficiency. They do not develop pituitary enlargement as is seen in patients with PROP1 mutations. Known genetic causes of isolated GH deficiency include inactivating mutations in the GHRHR gene and in the GHN gene. GHRH action is required for somatotrope proliferation (late in pituitary development) and for GH synthesis and secretion. A defective GHRHR impacts all these mechanisms, and affected patients have pituitary hypoplasia and severe isolated GH deficiency.35,36 The clinical phenotype is one of proportionate dwarfism with relative microcephaly, normal fertility, and normal lactation.36 Mutations in the GHN gene affect GH production directly.37 The GH locus is prone to deletions because of gene duplication, which includes homologous sequences in the regions flanking the duplicated genes. Deletions of 6.7, 7.0, and 7.6 kb are typical; affected homozygous patients have severe GH deficiency (termed type IA). A similar phenotype occurs with other severely inactivating mutations, such as nonsense and frameshift mutations. A milder form (termed type IB) is seen with less disabling mutations, such as missense mutations in which some, albeit abnormal, GH is produced. In type IA, secondary resistance to exogenous GH administration is seen frequently, but not always, because of the formation of high-titer anti-GH antibodies to the “foreign” GH protein. Patients with type IB, on the other hand, respond well to exogenous GH because they are immune tolerant to GH. A special case in this category is a bioinactive GH.38 Dominantly inherited GHN gene mutations (termed type II) are caused by splice-site mutations in one allele that result in skipping of exon 3, with the resultant abnormal GH protein exerting a deleterious (dominant negative) influence on the normal GH produced by the intact allele. Improper disulfide pairing/aggregation or deranged transport to secretory granules are postulated mechanisms. In one case, a mutant GH was shown to act as an antagonist at the level of the GHR.39 Yet another type of isolated GH deficiency is inherited in an X-linked manner (termed type III). The gene involved is unknown but resides close to the gene for “Bruton tyrosine kinase,” an enzyme that is crucial for B-lymphocyte function. Hence, this type of GH deficiency is usually associated with hypo- or agammaglobu-linemia. Interestingly, defects in the GHRH gene have not been identified to date, despite the fact that many cases of idiopathic GH deficiency are thought to be due to GHRH deficiency.
PROP1 gene, with resulting frameshift, is the most common cause.33 The clinical phenotype is one of familial dwarfism, with hypothyroidism and hypogonadism. Of interest, pituitary enlargement frequently occurs in affected patients—a finding whose pathogenesis is not clear. Mutations in the PIT1 gene are another cause of combined pituitary hormone deficiency.34 At least eight different mutations have been described; most are recessively inherited, but some are dominant negative (i.e., the abnormal gene product interferes with that derived from the normal allele). Pit-1 is important for development of somatotropes, lactotropes, and thyrotropes; affected patients suffer from GH, prolactin, and TSH deficiency. They do not develop pituitary enlargement as is seen in patients with PROP1 mutations. Known genetic causes of isolated GH deficiency include inactivating mutations in the GHRHR gene and in the GHN gene. GHRH action is required for somatotrope proliferation (late in pituitary development) and for GH synthesis and secretion. A defective GHRHR impacts all these mechanisms, and affected patients have pituitary hypoplasia and severe isolated GH deficiency.35,36 The clinical phenotype is one of proportionate dwarfism with relative microcephaly, normal fertility, and normal lactation.36 Mutations in the GHN gene affect GH production directly.37 The GH locus is prone to deletions because of gene duplication, which includes homologous sequences in the regions flanking the duplicated genes. Deletions of 6.7, 7.0, and 7.6 kb are typical; affected homozygous patients have severe GH deficiency (termed type IA). A similar phenotype occurs with other severely inactivating mutations, such as nonsense and frameshift mutations. A milder form (termed type IB) is seen with less disabling mutations, such as missense mutations in which some, albeit abnormal, GH is produced. In type IA, secondary resistance to exogenous GH administration is seen frequently, but not always, because of the formation of high-titer anti-GH antibodies to the “foreign” GH protein. Patients with type IB, on the other hand, respond well to exogenous GH because they are immune tolerant to GH. A special case in this category is a bioinactive GH.38 Dominantly inherited GHN gene mutations (termed type II) are caused by splice-site mutations in one allele that result in skipping of exon 3, with the resultant abnormal GH protein exerting a deleterious (dominant negative) influence on the normal GH produced by the intact allele. Improper disulfide pairing/aggregation or deranged transport to secretory granules are postulated mechanisms. In one case, a mutant GH was shown to act as an antagonist at the level of the GHR.39 Yet another type of isolated GH deficiency is inherited in an X-linked manner (termed type III). The gene involved is unknown but resides close to the gene for “Bruton tyrosine kinase,” an enzyme that is crucial for B-lymphocyte function. Hence, this type of GH deficiency is usually associated with hypo- or agammaglobu-linemia. Interestingly, defects in the GHRH gene have not been identified to date, despite the fact that many cases of idiopathic GH deficiency are thought to be due to GHRH deficiency.
 |
The majority of cases of congenital GH deficiency (isolated or combined) are sporadic; they are thought to be caused by birth trauma, cerebral insults, and congenital malformations or tumors affecting hypothalamic-pituitary development or function. Approximately 10% of such patients have abnormal magnetic resonance imaging (MRI) scans. In one family affected with two cases of septo-optic dysplasia, an inactivating mutation has been found in the gene encoding the early developmental transcription factor Hesx1, also known as Rpx (Rathke Pouch Homeobox).40 The incidence of GH deficiency is estimated as 1 per 4000 to 10,000 births.
Acquired Growth Hormone Deficiency.
GH deficiency can be acquired at any time during the life span, depending on when a hypothalamic-pituitary lesion or insult occurs. As already indicated, normal aging is associated with declining GH secretion during adulthood, and senescence resembles progressive GH insufficiency. During the growing years, the hallmark of GH deficiency is growth retardation; in adult life, the signs are more subtle changes in body composition and metabolism. Because of the lack of obvious manifestations, GH deficiency in adults is often not recognized, being diagnosed only after a long delay, or not considered clinically important. Moreover, only recently has GH deficiency in adults become amenable to therapy.
With pituitary lesions, the pattern of loss of pituitary hormones tends to follow a certain order: GH, gonadotropins, and prolactin tend to be lost early, whereas TSH and ACTH are affected later. This may be related to the abundance and/or spatial distribution of the respective cell types, with somato-tropes being the most prevalent (accounting for ˜50% of the anterior pituitary mass).
CLINICAL PRESENTATION AND DIAGNOSIS
In childhood, GH deficiency soon declares itself because of growth retardation. The GH dependence of somatic growth begins at or shortly after birth. If coexistent TSH deficiency is present, the resulting hypothyroidism contributes to and aggravates growth retardation. Organic causes for GH deficiency should be sought, but frequently no lesion is found, leading to the diagnosis of idiopathic GH deficiency. This entity is attributed to partial GHRH deficiency, although direct proof is
difficult to obtain. Patients with idiopathic GH deficiency usually have blunted, but not absent, GH responses to secretagogues. As already mentioned, the boundaries between normal, insufficient, and deficient GH secretion are ill defined because GH secretion rates are highly variable in normal people. This may render the diagnosis of idiopathic GH deficiency difficult. Growth retardation due to GH deficiency results in proportionate dwarfism and a progressive deviation from the normal growth curve. Untreated severe isolated GH deficiency results in adult heights of 120–140 cm in males and 110–130 cm in females. Other manifestations of GH deficiency during childhood are hypoglycemia, micropenis, and craniofacial abnormalities with saddle nose and facial hypoplasia. Patients tend to be “chubby” because of increased adipose tissue, particularly in the truncal area. Bone maturation is delayed by several years, and puberty is delayed by 2 to 3 years. Postpubertal men have a characteristic, high-pitched voice. The impact of GH deficiency on somatic growth and development is described in detail in Chapter 18, Chapter 92 and Chapter 198 Figure 12-13 shows the phenotype of adult patients with isolated GH deficiency. GH deficiency must be differentiated from other forms of short stature (constitutional, idiopathic, familial, etc.) that are much more common.
difficult to obtain. Patients with idiopathic GH deficiency usually have blunted, but not absent, GH responses to secretagogues. As already mentioned, the boundaries between normal, insufficient, and deficient GH secretion are ill defined because GH secretion rates are highly variable in normal people. This may render the diagnosis of idiopathic GH deficiency difficult. Growth retardation due to GH deficiency results in proportionate dwarfism and a progressive deviation from the normal growth curve. Untreated severe isolated GH deficiency results in adult heights of 120–140 cm in males and 110–130 cm in females. Other manifestations of GH deficiency during childhood are hypoglycemia, micropenis, and craniofacial abnormalities with saddle nose and facial hypoplasia. Patients tend to be “chubby” because of increased adipose tissue, particularly in the truncal area. Bone maturation is delayed by several years, and puberty is delayed by 2 to 3 years. Postpubertal men have a characteristic, high-pitched voice. The impact of GH deficiency on somatic growth and development is described in detail in Chapter 18, Chapter 92 and Chapter 198 Figure 12-13 shows the phenotype of adult patients with isolated GH deficiency. GH deficiency must be differentiated from other forms of short stature (constitutional, idiopathic, familial, etc.) that are much more common.
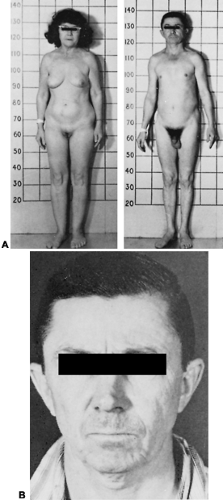 FIGURE 12-13. A, Two siblings with familial isolated growth hormone (GH) deficiency. Note the proportionate short stature and normal sexual development. B, The “child-like,” finely wrinkled face of a 41-year-old man with isolated GH deficiency. |
In adults, GH deficiency should be suspected when a pituitary or hypothalamic lesion is found, or when a history of childhood GH deficiency is present. Patients who had idiopathic GH deficiency during childhood may or may not be GH deficient as adults, so that retesting of GH secretion in adulthood is required. Approximately two-thirds of such patients are found to have normal GH secretion as adults.41 One explanation for this phenomenon is that GH secretion rates during childhood and particularly during puberty are higher than in adult life, and that a partial limitation in GH production may yield levels that are inadequate during the growth years but sufficient in adulthood. Patients with pituitary lesions should be tested for GH deficiency even in the absence of other pituitary hormone deficiencies because somatotrope dysfunction may be an early manifestation of pituitary failure. This has therapeutic consequences, because such patients may benefit from GH treatment. The clinical manifestations of the adult GH deficiency syndrome are listed in Table 12-4.
 |
Diagnosis relies on the demonstration of an underlying cause (a pituitary or hypothalamic lesion, a genetic defect, or a history of radiation), on provocative tests of GH secretion, and on IGF-I measurement. A low serum IGFBP-3 and a high IGFBP-2 level can be used as corroborative indicators. The typical, but arbitrary, cutoff for a normal peak GH level in response to provocative tests is 5 to 7 ng/mL in a polyclonal assay, and 2.5 to 3.5 ng/mL in a monoclonal assay. The Growth Hormone Research Society Consensus Guidelines for adult GH deficiency define a cutoff of 3 ng/mL in response to an insulin-tolerance test as “severe GH deficiency,” with a gray zone between 3 and 5 ng/mL, using a polyclonal assay.42 These cutoffs cannot be absolute because of the problems inherent in the assays mentioned above, and because of interindividual variations in GH response. Furthermore, tests that are less potent than the insulin-tolerance test may yield a lower GH response. Therefore, provocative tests must be interpreted in the clinical context. A finding of a low
serum IGF-I level is helpful in the absence of other causes for decreased IGF production, such as nutritional deprivation. However, difficulty arises because of the wide normal range for IGF-I levels. IGF-I levels are highly age dependent and must be judged against age-appropriate normative values. In very young children, in whom IGF-I levels are naturally low, differentiation of GH deficiency from normal production may be difficult based on the IGF-I level. In adults, particularly older adults, considerable overlap also exists between the normal IGF-I range and that seen in GH deficiency (Fig. 12-14).
serum IGF-I level is helpful in the absence of other causes for decreased IGF production, such as nutritional deprivation. However, difficulty arises because of the wide normal range for IGF-I levels. IGF-I levels are highly age dependent and must be judged against age-appropriate normative values. In very young children, in whom IGF-I levels are naturally low, differentiation of GH deficiency from normal production may be difficult based on the IGF-I level. In adults, particularly older adults, considerable overlap also exists between the normal IGF-I range and that seen in GH deficiency (Fig. 12-14).
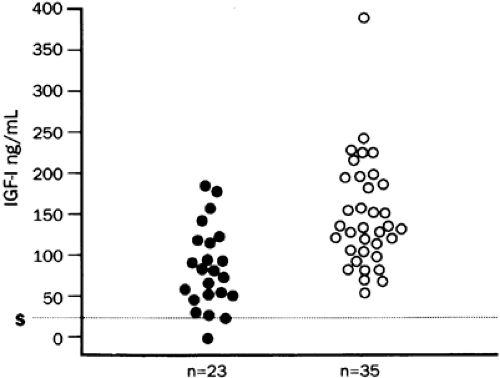 FIGURE 12-14. Serum insulin-like growth factor-I (IGF-I) levels in adult hypopituitary patients, aged 17 to 77 years (closed circles), compared to those in age-matched normal controls (open circles). All patients had growth hormone deficiency, as assessed by the insulin-tolerance test, and deficiency of other pituitary hormones. Note the overlap between normal and hypopituitary IGF-I levels. (S, assay sensitivity for IGF-I [25 ng/mL]; n, number of subjects tested.) (Adapted from Hoffman DM, O’Sullivan AJ, Baxter RC, Ho KKY. Diagnosis of growth-hormone deficiency in adults. Lancet 1994; 343:1064.)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
