INHERITED DISORDERS OF VITAMIN D METABOLISM AND ACTIVITY
Autosomal recessive 1α -hydroxylase deficiency (also known as hereditary pseudovitamin D deficiency rickets or vitamin D–dependent rickets type I) is an autosomal-recessive disorder in which the renal 25(OH)D 1α-hydroxylase that converts 25(OH)D to 1,25(OH)2D is dysfunctional. Individuals with this disorder have a mutation in the gene encoding a specific cytochrome P450 that donates electrons to the substrate, 25(OH)D.14 Thus, this enzyme component is essential to the hydroxylation reaction. Onset of symptoms is usually within 4 to 12 months of age, and the clinical course and biochemical features are similar to those of severe vitamin D–deficiency rickets. In contrast to vitamin D–deficient rickets, there is a history of adequate dietary intake of vitamin D or of sunlight exposure. In untreated patients, the serum 25(OH)D level is normal. The serum 1,25(OH)2D is low to low-normal and remains so in patients treated with pharmacologic doses of vitamin D or 25(OH)D. Patients with 1α-hydroxylase deficiency may respond to high dosages of vitamin D or 25(OH)D; however, treatment with 1,25(OH)2D is optimal, using initial dosages from 0.5 to 3.0 μg per day and maintenance dosages ranging from 0.25 to 2.0 μg per day.
Hereditary vitamin D resistance (also known as vitamin D– dependent rickets type II) is a rare inherited disorder transmitted as an autosomal-recessive trait. The clinical, radiologic, and most of the biochemical features are similar to those observed in 1α-hydroxylase deficiency (Fig. 70-5). The major biochemical distinction is that circulating 1,25(OH)2D levels are high in patients with hereditary vitamin D resistance and low in patients with 1α-hydroxylase deficiency. A unique clinical feature in some kindreds with hereditary vitamin D resistance is total body and scalp alopecia. Other patients may demonstrate oligodontia (Fig. 70-6). Studies in affected individuals of several kindreds have revealed defects in the 1,25(OH)2D receptor, which is one of a large superfamily of hormone receptors, including glucocorticoid, mineralocorticoid, and thyroid hormone receptors. Mutations, which have been identified in the gene encoding this receptor, result in altered DNA binding, altered ligand binding, or interruption of complete synthesis of the receptor (see Chap. 54). In some families, no genetic defect has been identified.
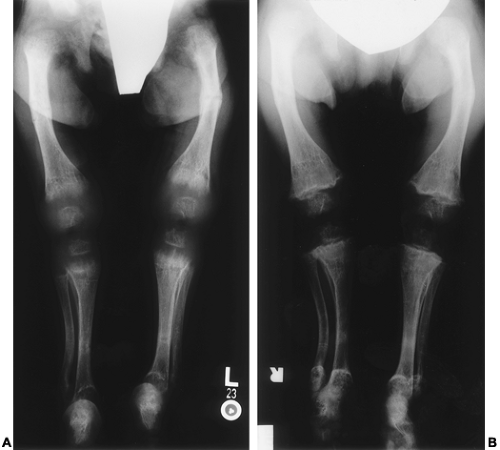 FIGURE 70-5. Rachitic changes in the lower extremities of a 2½-year-old girl with hereditary resistance to vitamin D. At presentation (A) severely deformed epiphyses, demineralization, and bilateral fractures of the femora and tibiae are evident. After 4 months of therapy with high-dose calcitriol (B), dramatic remodeling of the metaphyseal edges has occurred. |
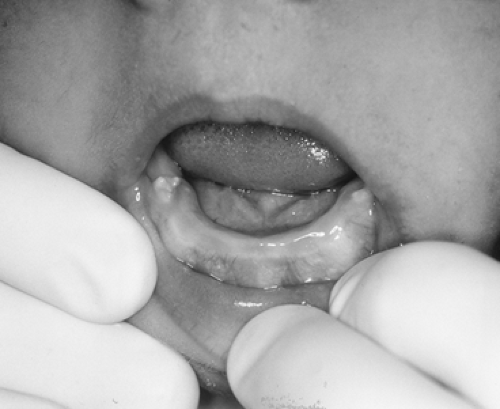 FIGURE 70-6. The limited eruption of teeth (oligodontia), as shown here, is characteristic of certain kindreds with hereditary resistance to vitamin D. |
Patients with hereditary vitamin D resistance have been treated with megadoses of vitamin D or vitamin D metabolites with variable responses to such therapy. Parenteral administration of calcium has been shown to completely correct the skeletal abnormalities in severe forms of this disorder.15
X-LINKED HYPOPHOSPHATEMIC RICKETS
X-Linked hypophosphatemic rickets16 (XLH) is inherited as an X-linked dominant trait and is characterized by hypophosphatemia and decreased renal tubular phosphate reabsorption. The inherited disorder may vary in degree, from severe to mild bone disease associated with hypophosphatemia, to hypophosphatemia alone without evidence of active or former rickets. The severity of bone disease does not correlate with the degree of hypophosphatemia. Children with XLH are usually seen in the second or third year of life with bowed legs and short stature. Controversy exists whether the mineralized tissue abnormalities are more severe in affected males compared with females.17,18 Hallmark laboratory findings include hypophosphatemia, normocalcemia, and elevated serum alkaline phosphatase activity. The tubular reabsorption rate and the maximum reabsorption rate of phosphate are decreased. The serum PTH level is usually normal but can be elevated in the untreated state. Hyperparathyroidism is not infrequently seen as a complication of therapy. In the untreated state, the serum 25(OH)D levels are normal. The serum 1,25(OH)2D levels are either normal or somewhat decreased. The “normal” circulating levels of this metabolite are inappropriately low in the presence of hypophosphatemia.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


