Normal red cell destruction
Red cell destruction normally occurs after a mean lifespan of 120 days when the cells are removed extravascularly by the macrophages of the reticuloendothelial (RE) system, especially in the marrow but also in the liver and spleen. As the cells have no nucleus, red cell metabolism gradually deteriorates as enzymes are degraded and not replaced and the cells become non-viable. The breakdown of haem from red cells liberates iron for recirculation via plasma transferrin mainly to marrow erythroblasts, and protoporphyrin which is broken down to bilirubin. Bilirubin circulates to the liver where it is conjugated to glucuronides which are excreted into the duodenum via bile and converted to stercobilinogen and stercobilin (excreted in faeces) (Fig. 6.1). Stercobilinogen and stercobilin are partly reabsorbed and excreted in urine as urobilinogen and urobilin. Globin chains are broken down to amino acids which are reutilized for general protein synthesis in the body.
Figure 6.1 (a) Normal red blood cell (RBC) breakdown. This takes place extravascularly in the macrophages of the reticuloendothelial system. (b) Intravascular haemolysis occurs in some pathological disorders.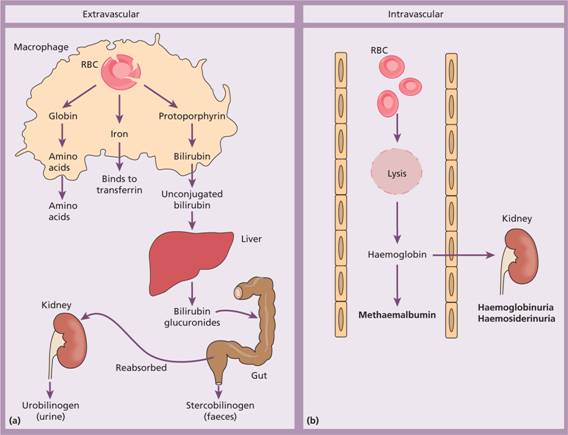
Haptoglobins are proteins present in normal plasma capable of binding haemoglobin. The haemoglobin–haptoglobin complex is removed from plasma by the RE system. Intravascular haemolysis (breakdown of red cells within blood vessels) plays little or no part in normal red cell destruction.
Introduction to haemolytic anaemias
Haemolytic anaemias are defined as those anaemias that result from an increase in the rate of red cell destruction. Because of erythropoietic hyperplasia and anatomical extension of bone marrow, red cell destruction may be increased several-fold before the patient becomes anaemic–compensated haemolytic disease. The normal adult marrow, after full expansion, is able to produce red cells at 6–8 times the normal rate provided this is ‘effective’. Therefore, haemolytic anaemia may not be seen until the red cell lifespan is less than 30 days. It leads to a marked reticulocytosis, particularly in the more anaemic cases.
Classification
Table 6.1 is a simplified classification of the haemolytic anaemias. Hereditary haemolytic anaemias are the result of ‘intrinsic’ red cell defects whereas acquired haemolytic anaemias are usually the result of an ‘extracorpuscular’ or ‘environmental’ change. Paroxysmal nocturnal haemoglobinuria (PNH) is the exception because although it is an acquired disorder, the PNH red cells have an intrinsic defect.
Table 6.1 Classification of haemolytic anaemias.
| Hereditary | Acquired |
| Membrane | Immune |
| Hereditary spherocytosis, hereditary elliptocytosis | Autoimmune |
| Metabolism | Warm antibody type |
| G6PD deficiency, pyruvate kinase deficiency | Cold antibody type |
| Haemoglobin | Alloimmune |
| Genetic abnormalities (Hb S, Hb C, unstable); see Chapter 7 | Haemolytic transfusion reactions |
| Haemolytic disease of the newborn | |
| Allografts, especially stem cell transplantation | |
| Drug associated | |
| Red cell fragmentation syndromes | |
| See Table 6.6 | |
| March haemoglobinuria | |
| Infections | |
| Malaria, clostridia | |
| Chemical and physical agents | |
| Especially drugs, industrial/domestic substances, burns | |
| Secondary | |
| Liver and renal disease | |
| Paroxysmal nocturnal haemoglobinuria |
G6PD, glucose-6-phosphate dehydrogenase; Hb, haemoglobin.
Clinical features
The patient may show pallor of the mucous membranes, mild fluctuating jaundice and splenomegaly. There is no bilirubin in urine but this may turn dark on standing because of excess urobilinogen. Pigment (bilirubin) gallstones may complicate the condition (Fig. 6.2) and some patients (particularly with sickle cell disease) develop ulcers around the ankle. Aplastic crises may occur, usually precipitated by infection with parvovirus which ‘switches off’ erythropoiesis, and are characterized by a sudden increase in anaemia and drop in reticulocyte count (see Fig. 22.4).
Figure 6.2 Ultrasound of multiple small pigment gallstones typical of those associated with hereditary spherocytosis. (Courtesy of Dr P. Wylie.)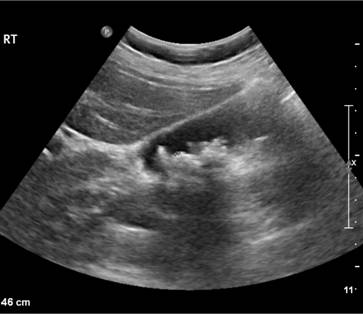
Rarely, folate deficiency may cause an aplastic crisis in which the bone marrow is megaloblastic.
Laboratory findings
The laboratory findings are conveniently divided into three groups.
1 Features of increased red cell breakdown:
(a) Serum bilirubin raised, unconjugated and bound to albumin;
(b) Urine urobilinogen increased;
(c) Serum haptoglobins absent because the haptoglobins become saturated with haemoglobin and the complex is removed by RE cells.
2 Features of increased red cell production:
(a) Reticulocytosis;
(b) Bone marrow erythroid hyperplasia; the normal marrow myeloid: erythroid ratio of 2 : 1 to 12 : 1 is reduced to 1 : 1 or reversed.
3 Damaged red cells:
(a) Morphology (e.g. microspherocytes, elliptocytes, fragments);
(b) Osmotic fragility, autohaemolysis, etc.;
(c) Specific enzyme, protein or DNA tests.
Intravascular and extravascular haemolysis
There are two mechanisms whereby red cells are destroyed in haemolytic anaemia. There may be excessive removal of red cells by macrophages of the RE system (extravascular haemolysis) or they may be broken down directly in the circulation (intravascular haemolysis) (Table 6.2; Fig. 6.1). Whichever mechanism dominates will depend on the pathology involved. In intravascular haemolysis, free haemoglobin is released which rapidly saturates plasma haptoglobins and the excess free haemoglobin is filtered by the glomerulus. If the rate of haemolysis saturates the renal tubular reabsorptive capacity, free haemoglobin enters urine (Fig. 6.3a) and, as iron is released, the renal tubules become loaded with haemosiderin. Methaemalbumin is also formed from the process of intravascular haemolysis.
Table 6.2 Causes of intravascular haemolysis.
| Mismatched blood transfusion (usually ABO) |
| G6PD deficiency with oxidant stress |
| Red cell fragmentation syndromes |
| Some autoimmune haemolytic anaemias |
| Some drug-and infection-induced haemolytic anaemias |
| Paroxysmal nocturnal haemoglobinuria |
| March haemoglobinuria |
| Unstable haemoglobin |
G6PD, glucose-6-phosphate dehydrogenase.
Figure 6.3 (a) Progressive urine samples in an acute episode of intravascular haemolysis showing haemoglobinuria of decreasing severity. (b) Prussian blue-positive deposits of haemosiderin in a urine spun deposit (Perls’ stain).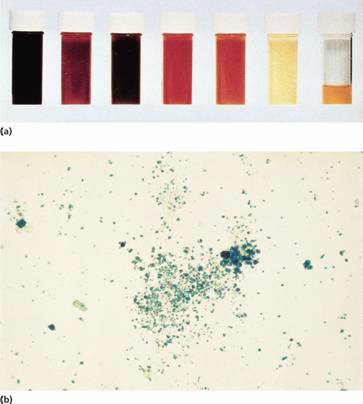
The main laboratory features of intravascular haemolysis are:
1 Haemoglobinaemia and haemoglobinuria;
2 Haemosiderinuria (iron storage protein in the spun deposit of urine (Fig. 6.3b));
3 Methaemalbuminaemia (detected spectrophotometrically by Schumm’s test).
Hereditary haemolytic anaemias
Membrane defects
Hereditary spherocytosis
Hereditary spherocytosis (HS) is the most common hereditary haemolytic anaemia in northern Europeans.
Pathogenesis
HS is usually caused by defects in the proteins involved in the vertical interactions between the membrane skeleton and the lipid bilayer of the red cell (Table 6.3; see Fig. 2.12). The loss of membrane may be caused by the release of parts of the lipid bilayer that are not supported by the skeleton. In HS, the marrow produces red cells of normal biconcave shape but these lose membrane and become increasingly spherical (loss of surface area relative to volume) as they circulate through the spleen and the rest of the RE system. Ultimately, the spherocytes are unable to pass through the splenic microcirculation where they die prematurely.
Table 6.3 Molecular basis of hereditary spherocytosis and elliptocytosis.
| Hereditary spherocytosis |
| Ankyrin deficiency or abnormalities |
| α-or β- spectrin deficiency or abnormalities |
| Band 3 abnormalities |
| Pallidin (protein 4.2) abnormalities |
| Hereditary elliptocytosis |
| α- or β-spectrin mutants leading to defective spectrin dimer formation |
| α- or β-spectrin mutants leading to defective spectrin–ankyrin associations |
| Protein 4.1 deficiency or abnormality |
| South-East Asian ovalocytosis (band 3 deletion) |
Clinical features
The inheritance is autosomal dominant with variable expression; rarely it may be autosomal recessive. The anaemia can present at any age from infancy to old age. Jaundice is typically fluctuating and is particularly marked if the haemolytic anaemia is associated with Gilbert’s disease (a defect of hepatic conjugation of bilirubin); splenomegaly occurs in most patients. Pigment gallstones are frequent (Fig. 6.2); aplastic crises, usually precipitated by parvovirus infection, may cause a sudden increase in severity of anaemia (see Fig. 22.5).
Haematological findings
Anaemia is usual but not invariable; its severity tends to be similar in members of the same family. Reticulocytes are usually 5–20%. The blood film shows microspherocytes (Fig. 6.4a) that are densely staining with smaller diameters than normal red cells.
Figure 6.4 (a) Blood film in hereditary spherocytosis. The spherocytes are deeply staining and of small diameter. Larger polychromatic cells are reticulocytes (confirmed by supravital staining). (b) Blood film in hereditary elliptocytosis.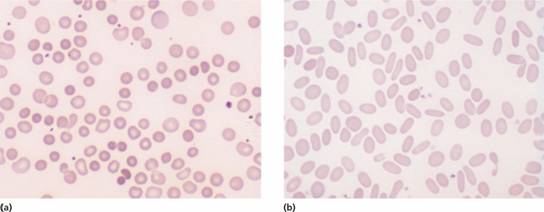
Investigation and treatment
A rapid fluorescent flow analysis of eosin-maleimide bound to red cells is used as a test for HS and membrane band 3 protein deficiency (Fig. 6.5). This has replaced the classic osmotic fragility test which showed the HS red cells to be excessively fragile in dilute saline solution. The identification of the exact molecular defect is not needed for management. The direct antiglobulin (Coombs) test is normal, excluding an autoimmune cause of spherocytosis and haemolysis.
Figure 6.5 Eosin-5-maleimide staining in hereditary spherocytosis (HS) showing reduced mean channel fluorescence due to membrane band 3 protein deficiency. (Courtesy of Mr G. Ellis.)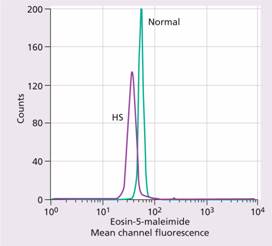
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


