Arterial thrombosis
Pathogenesis
Atherosclerosis of the arterial wall, plaque rupture and endothelial injury expose blood to subendothelial collagen and tissue factor. This initiates the formation of a platelet nidus on which platelets adhere and aggregate.
Platelet deposition and thrombus formation are important in the pathogenesis of atherosclerosis. Platelet-derived growth factor stimulates the migration and proliferation of smooth muscle cells and fibroblasts in the arterial intima. Regrowth of endothelium and repair at the site of arterial damage and incorporated thrombus result in thickening of the vessel wall.
As well as blocking arteries locally, emboli of platelets and fibrin may break away from the primary thrombus to occlude distal arteries. Examples are carotid artery thrombi leading to cerebral thrombosis and transient ischaemic attacks and heart valve and chamber thrombi leading to systemic emboli and infarcts (Fig. 27.1).
Figure 27.1 Arteriogram showing saddle embolus at the aortic bifurcation (dotted arrow) and embolus in the left common iliac artery (solid arrow).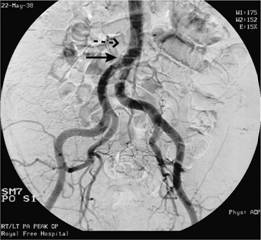
Clinical risk factors
The risk factors for arterial thrombosis are related to the development of atherosclerosis and are listed in Table 27.1. The identification of patients at risk is largely based on clinical assessment. A number of epidemiological studies have resulted in the construction of coronary artery thrombosis risk profiles based on sex, age, elevated blood pressure, high levels of serum cholesterol, glucose intolerance, cigarette smoking and electrocardiogram abnormalities. These profiles have allowed pre-symptomatic assessment of young and apparently fit subjects and are valuable in counselling a change in lifestyle or for recommending medical therapy in individuals at risk. Epidemiological evidence implicates four inflammatory markers, fibrinogen, C-reactive protein, lipoprotein-associated phospholipase A2 and interleukin-6 as predictive for coronary heart disease.
Table 27.1 Risk factors for arterial thrombosis (atherosclerosisis).
| Positive family history |
| Male sex |
| Hyperlipidaemia |
| Hypertension |
| Diabetes mellitus |
| Gout |
| Polycythaemia |
| Hyperhomocysteinaemia |
| Cigarette smoking |
| ECG abnormalities |
| Elevated CRP, IL6, fibrinogen, lipoprotein-associated phospholipase A2 |
| Lupus anticoagulant |
| Collagen vascular diseases |
| Behçet’s disease |
CRP, C-reactive protein; ECG, electrocardiogram.
Pathogenesis and risk factors
Virchow’s triad suggests that there are three components that are important in thrombus formation:
1 Slowing down of blood flow;
2 Hypercoagulability of the blood; and
3 Vessel wall damage.
For venous thrombosis, increased systemic coagulability and stasis are most important, vessel wall damage being less important than in arterial thrombosis, although it may be important in patients with sepsis and indwelling catheters. Stasis allows the completion of blood coagulation at the site of initiation of the thrombus (e.g. behind the valve pockets of the leg veins in immobile patients).
Table 27.2 lists a number of recognized risk factors.
Table 27.2 Hereditary and acquired risk factors for venous thrombosis.
| Hereditary haemostatic disorders |
| Factor V Leiden |
| Prothrombin G20210A variant |
| Protein C deficiency |
| Antithrombin deficiency |
| Protein S deficiency |
| Dysfibrinogenaemia |
| ABO blood group |
| Hereditary or acquired haemostatic disorders |
| Raised plasma levels of factor VIII |
| Raised plasma levels of fibrinogen |
| Raised plasma levels of homocysteine |
| Acquired disorders |
| Lupus anticoagulant |
| Oestrogen therapy (oral contraceptive and HRT) |
| Heparin-induced thrombocytopenia |
| Pregnancy and puerperium |
| Surgery, especially abdominal, hip and knee surgery |
| Major trauma |
| Malignancy |
| Acutely ill hospitalized medical patients including cardiac or respiratory failure, infection, inflammatory bowel disorders |
| Myeloproliferative disease |
| Hyperviscosity, polycythaemia |
| Stroke |
| Pelvic obstruction |
| Nephrotic syndrome |
| Dehydration |
| Varicose veins |
| Age |
| Obesity |
| Paroxysmal nocturnal haemoglobinuria |
| Behçet’s disease |
HRT, hormone replacement therapy.
Hereditary disorders of haemostasis
The prevalence of inherited disorders associated with increased risk of thrombosis is higher than that of hereditary bleeding disorders. A hereditary ‘thrombophilia’ should be particularly suspected in young patients who develop spontaneous thrombosis, recurrent DVTs (Fig. 27.2) or an unusual site of thrombosis (e.g. axillary, splanchnic veins, sagittal sinus). Several abnormalities are now well characterized (Table 27.2).
Figure 27.2 Diagnostic imaging of deep vein thrombosis (DVT) and pulmonary embolus (PE). (a) Colour power Doppler ultrasound of the right femoral vessels with compression shows normal flow in the femoral artery but absent flow in the vein because of thrombus. A normal vein would collapse with compression of the probe. (Courtesy of Dr Tony Young.) (b) Femoral venogram demonstrating extensive thrombus within the right external iliac vein. (Courtesy of Dr I.S. Francis and Dr A.F. Watkinson.) (c) Computed tomography (CT) pulmonary angiography: a coronal image shows bilateral filling defects (green crosses) in the central central pulmonary arteries indicating pulmonary emboli. (Courtesy of Dr Tony Young.)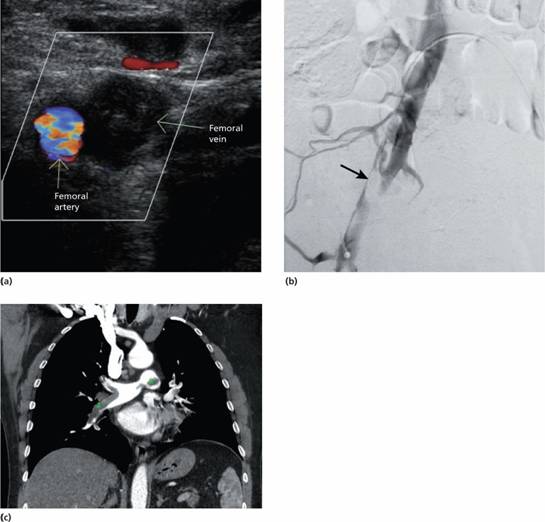
Factor V Leiden gene mutation (activated protein C resistance)
This is the most common inherited cause of an increased risk of venous thrombosis. It occurs in approximately 3–7% of Caucasian factor V alleles. There is failure of activated protein C (APC) when added to plasma to prolong the activated partial thromboplastin time (APTT) test. Protein C, when activated, breaks down activated factor V so APC should slow the clotting reaction and prolong the APTT. APC resistance is caused by a genetic polymorphism in the factor V gene (replacement of arginine at position 506 with glutamine–Arg506Gln) which makes factor V less susceptible to cleavage by APC (Fig. 27.3). This is called the factor V Leiden mutation. The frequency of factor V Leiden in the general population in Western countries means that it cannot be regarded as a rare mutation but as a genetic polymorphism that is maintained in the population (Fig. 27.4). Presumably, individuals with this allele have been ‘selected’, probably because of a reduced bleeding tendency (e.g. post-partum). It does not increase the risk of arterial thrombosis.
Figure 27.3 The genetic basis of factor V Leiden. (a) Activated protein C (APC) inactivates factor Va by proteolytic cleavage at three sites in the Va heavy chain. (b) In the factor V Leiden mutation the Arg506Gln polymorphism leads to glutamine at position 506 with less efficient inactivation of factor V by APC and increased risk of thrombosis.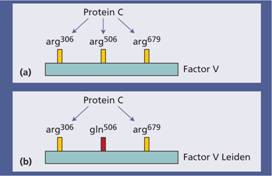
Figure 27.4 The incidence of carriers of factor V Leiden in different countries.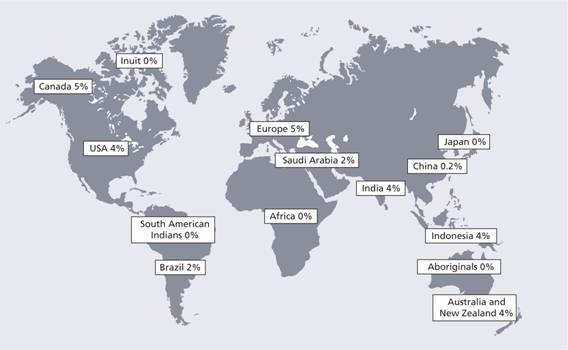
Patients who are heterozygous for factor V Leiden are at an approximately five- to eight-fold increased risk of venous thrombosis compared to the general population but only 10% of carriers develop thrombosis during their lifetime. Individuals who are homozygous have a 30–140- fold risk. Following venous thrombosis they have a higher risk of re-thrombosis than individuals with DVT but normal factor V.
The incidence of factor V Leiden in patients with venous thrombosis is approximately 20–40%. Polymerase chain reaction (PCR) screening for the mutation is relatively simple and the test is widely performed. The absolute risk of thrombosis will depend on many other factors and it is difficult to advise individual patients of their risk. At present it is not recommended to start anticoagulation therapy in individuals with the Leiden mutation, even if homozygous, with no history of thrombosis. A small minority of patients with APC resistance do not have factor V Leiden but have other mutations of factor V.
Antithrombin deficiency
Inheritance is autosomal dominant. There are recurrent venous thromboses usually starting in early adult life. Arterial thrombi occur occasionally. Antithrombin concentrates are available and are used to prevent thrombosis during surgery or childbirth. Many molecular variants of antithrombin have been categorized and are associated with varying degrees of risk of thrombosis.
Protein C deficiency
Inheritance is autosomal dominant with variable penetrance. Protein C levels in heterozygotes are approximately 50% of normal. Characteristically, many patients develop skin necrosis as a result of dermal vessel occlusion when treated with warfarin, thought to be caused by reduction of protein C levels even further in the first day or two of warfarin therapy before reduction in the levels of the vitamin K-dependent clotting factors, especially factors II and X. Rarely, infants may be born with homozygous deficiency and characteristically present with severe disseminated intravascular coagulation (DIC) or purpura fulminans in infancy. APC concentrates are available and are used in selected acquired cases of severe sepsis with DIC as well as in genetic protein C deficiency. APC is also used in selected patients with multiorgan failure and sepsis for its anti-inflammitory, anticoagulant and pro-fibrinolytic effects.
Protein S deficiency
Protein S deficiency has been found in a number of families with a thrombotic tendency. It is a cofactor for protein C and the clinical features are similar to protein C deficiency, including a tendency to skin necrosis with warfarin therapy. The inheritance is autosomal dominant.
Prothrombin allele G20210A
Prothrombin allele G20210A is a variant (prevalence 2–3% in the population) that leads to increased plasma prothrombin levels and increases thrombotic risk by fivefold. It is probable that the cause of venous thrombosis with this mutation and with high levels of factors VIII, IX and XI is that sustained generation of thrombin results in pro longed down-regulation of fibrinolysis through activation of thrombin-activated fibrinolysis inhibitor (see p. 325).
Hyperhomocysteinaemia
Higher levels of plasma homocysteine may be genetic or acquired and are associated with increased risk for both venous and arterial thrombosis. However, recent large trials show little evidence that lowering the levels reduces these risks.
Homocysteine is derived from dietary methionine and is removed by either remethylation to methionine or conversion to cysteine via a trans-sulphuration pathway (Fig. 27.5). Classic homocystinuria is a rare autosomal recessive disorder caused by deficiency of cystathione β-synthase, the enzyme responsible for trans-sulphuration. Vascular disease and thrombosis are major features of the disease. Heterozygous cystathione β-synthase deficiency is present in approximately 0.5% of the population and leads to a moderate increase in homocysteine. Methylene tetrahydrofolate reductase is involved in the remethylation pathway and a common thermolabile variant of the enzyme may be responsible for mild homocysteinaemia (above 15μ mol/L) although this may only be seen in the presence of folate or vitamin B12 deficiency. Acquired risk factors for hyperhomocysteinaemia include deficiencies of folate, vitamin B12 or vitamin B6, drugs (e.g. ciclosporin), renal damage and smoking. The levels also increase with age and are higher in men and post-menopausal females.
Figure 27.5 The metabolism of homocysteine. Homocysteine is derived from dietary methionine and is metabolized either by the trans-sulphuration or remethylation pathways. Trans-sulphuration proceeds using the cystathionine β-synthase (CBS) enzyme with vitamin B6 as a cofactor. Remethylation involves the action of methionine synthase (MS) on 5-methyl-THF with vitamin B12 as a cofactor. In addition, methylene-tetrahydrofolate reductase (MTHFR) is involved in this cycle.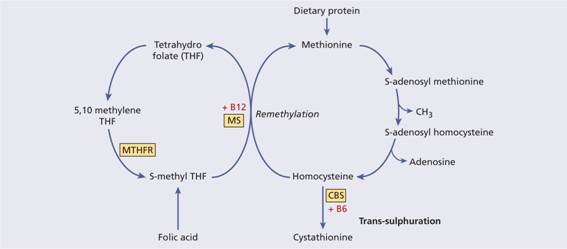
Defects of fibrinogen
Defects of fibrinogen are usually clinically silent or cause excess bleeding. Thrombosis is a rare association.
ABO blood group
Non-O blood group carriers have a higher risk of venous thrombosis or embolism than O carriers. This is related to their higher plasma levels of von Willebrand factor and factor VIII.
Hereditary or acquired disorders of haemostasis
High factor VIII or fibrinogen levels are also associated with venous thrombosis. The combination of multiple risk factors is associated with increased risk of thrombosis. If these are persistent they may represent a reason for extended anticoagulation.
Acquired risk factors
These may cause thrombosis in patients without another identifiable abnormality but are more likely to do so if an inherited predisposing abnormality (e.g. factor V Leiden) is also present.
Postoperative venous thrombosis
This is more likely to occur in the elderly, obese, those with a previous or family history of venous thrombosis, and in those in whom major abdominal or hip operations are performed. Elasticated stockings or mechanical methods (see p. 377) are used to reduce the risk of DVT.
Venous stasis and immobility
These factors are probably responsible for the high incidence of postoperative venous thrombosis and for venous thrombosis associated with congestive cardiac failure, myocardial infarction and varicose veins. In atrial fibrillation, thrombin generation from accumulation of activated clotting factors leads to a high risk clot formation in the atrial appendage and consequent systemic embolization. The use of muscle relaxants during anaesthesia may also contribute to venous stasis. Venous thrombosis also has a higher frequency after prolonged aeroplane journeys.
Malignancy
Patients with carcinoma of the ovary, brain and pancreas have a particularly increased risk of venous thrombosis but there is an increased risk with all cancers. The tumours produce tissue factor and a procoagulant that directly activates factor X. Mucin-secreting adenocarcinomas may be associated with DIC.
Inflammation
This up-regulates procoagulant factors, down-regulates anticoagulant pathways, particularly protein C. Thrombosis is particularly likely in inflammatory bowel disease, Behçet’s disease, systemic tuberculosis, systemic lupus erythematosus (SLE) and diabetes.
Blood disorders
Increased viscosity, thrombocytosis, altered platelet membrane receptors and responses are possible factors for the high incidence of thrombosis in patients with polycythaemia vera and essential thrombocythaemia. Testing for the JAK2 V617F mutation may indicate an otherwise unsuspected myeloproliferative disease in patients with hepatic or portal vein thrombosis. There is a high incidence of venous thrombosis, including thrombi in large veins (e.g. the hepatic vein) in patients with paroxysmal nocturnal haemoglobinuria. An increased tendency to venous thrombosis has been observed in patients with sickle cell disease, with post-splenectomy thrombocytosis and those with a paraprotein.
Oestrogen therapy
Oestrogen therapy, particularly high dose therapy, is associated with increased plasma levels of factors II, VII, VIII, IX and X and depressed levels of antithrombin and tissue plasminogen activator in the vessel wall. There is a high incidence of postoperative venous thrombosis in women on high dose oestrogen therapy and full dose oestrogen-containing oral contraceptives. The risk is much less with low dose oestrogen contraceptive preparations. Hormone replacement therapy also increases the risk of thrombosis, largely obviated by the use of low oestrogen preparations.
The antiphospholipid syndrome
The antiphospholipid syndrome (APS) can be defined as the occurrence of venous and arterial thrombosis and/or recurrent miscarriage in association with laboratory evidence of persistent antiphospholipid antibody. The predominant antibodies in this disorder are directed against protein antigens that bind to anionic phospholipids, such as β2-glycoprotein 1 (β2-GPI-1) and prothrombin.
One antiphospholipid antibody is the ‘lupus anticoagulant’ (LA) which was initially detected in patients with SLE, and is identified by a prolonged plasma APTT which does not correct with a 50: 50 mixture of normal plasma. Paradoxically, in view of its name, it is associated with venous and arterial thrombosis. A second test dependent on limiting quantities of phospholipid (e.g. the dilute Russell’s viper venom test) is also used in diagnosis. Whereas lupus anticoagulants are reactive in the fluid phase, other antiphospholipid antibodies, such as anticardiolipin antibodies and antibodies to β2-GPI-1, are identified by solid phase immunoassay. Both solid phase assays and coagulation tests for LA should be used in the diagnosis of APS.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


