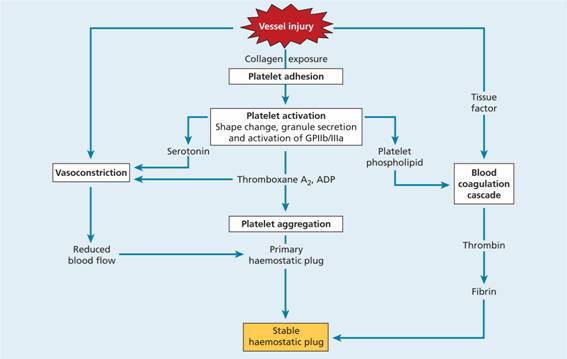
An efficient and rapid mechanism for stopping bleeding from sites of blood vessel injury is clearly essential for survival. Nevertheless, such a response needs to be tightly controlled to prevent extensive clots developing and to break down such clots once damage is repaired. The haemostatic system thus represents a delicate balance between procoagulant and anticoagulant mechanisms allied to a process for fibrinolysis. The five major components involved are platelets, coagulation factors, coagulation inhibitors, fibrinolysis and blood vessels. These are described later in the haemostatic response section on p. 324.
Components of the haemostatic response
Platelets
Platelet production
Platelets are produced in the bone marrow by fragmentation of the cytoplasm of megakaryocytes, one of the largest cells in the body. The precursor of the megakaryocyte–the megakaryoblast–arises by a process of differentiation from the haemopoietic stem cell (see Fig. 1.2). The megakaryocyte matures by endomitotic synchronous replication (i.e. DNA replication in the absence of nuclear or cytoplasmic division) enlarging the cytoplasmic volume as the number of nuclear lobes increase in multiples of two (Fig. 24.2). Very early on invaginations of plasma membrane are seen, called the demarcation membrane, which evolves through the development of the megakaryocyte into a highly branched network. At a variable stage in development, most commonly at the eight nucleus stage, the cytoplasm becomes granular. Mature megakaryocytes are extremely large, with an eccentric placed single lobulated nucleus and a low nuclear: cytoplasmic ratio (Fig. 24.3). Platelets form by fragmentation from the tips of cytoplasmic extensions of megakaryocyte cytoplasm, each megakaryocyte giving rise approximately to 1000–5000 platelets (Fig. 24.3c). The time interval from differentiation of the human stem cell to the production of platelets averages 10 days.
Figure 24.2 Simplified diagram to illustrate platelet production from megakaryocytes.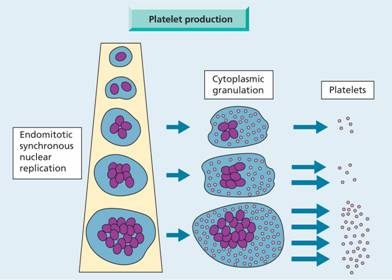
Figure 24.3 Megakaryocytes: (a) immature form with basophilic cytoplasm; (b) mature form with many nuclear lobes and pronounced granulation of the cytoplasm. (c) Megakaryocyte in culture, stained for α-tubulin (green). Proplatelets can be seen budding from the tips of megakaryocyte cytoplasm. (From Pecci A. et al. (2009) Thrombosis and Haemostasis 109, 90–96, with permission; with thanks to Professor Pecci for Fig. 24.3 c.)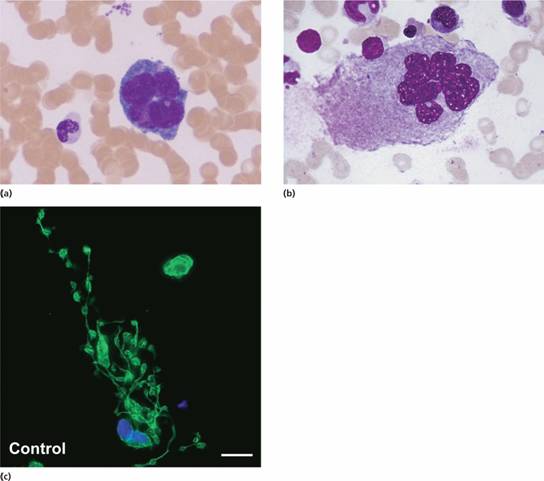
Thrombopoietin is the major regulator of platelet production and is constitutively produced by the liver and kidneys. Thrombopoietin increases the number and rate of maturation of megakaryocytes via c-MPL receptor. Platelet levels start to rise 6 days after the start of therapy and remain high for 7–10 days. Although thrombopoietin itself is not available for clinical use, thrombomimetic agents which bind to c-MPL are now used clinically to increase the platelet count (see p. 336). Platelets also have c-MPL receptors for thrombopoietin and remove it from the circulation. Therefore, levels are high in thrombocytopenia as a result of marrow aplasia but low in patients with raised platelet counts.
The normal platelet count is approximately 250 × 109/L (range 150–400 × 109/L) and the normal platelet lifespan is 7–10 days. This is determined by the ratio of the apoptotic BAX and anti-apopotic BCL-2 proteins in the cell. Up to one-third of the marrow output of platelets may be trapped at any one time in the normal spleen but this rises to 90% in cases of massive splenomegaly (see Fig. 25.9).
Platelet structure
Platelets are extremely small and discoid, 3.0 × 0.5 μ m in diameter, with a mean volume of 7–11 fL. The ultrastructure of platelets is represented in Figure 24.4. The glycoproteins of the surface coat are particularly important in the platelet reactions of adhesion and aggregation which are the initial events leading to platelet plug formation during haemostasis. Adhesion to collagen is facilitated by glycoprotein Ia (GPIa). Glycoproteins Ib (defective in Bernard–Soulier syndrome) and IIb/IIIa (also called α IIb and β3) (defective in Glanzmann ’ s thrombasthenia) are important in the attachment of platelets to von Willebrand factor (VWF) and hence to vascular subendothelium (Fig. 24.5) where signalling interactions occur (Fig. 24.6). The binding site for IIb/IIIa is also the receptor for fibrinogen which is important in platelet–platelet aggregation.
Figure 24.4 The ultrastructure of platelets. ADP, adenosine diphosphate; PDGF, platelet-derived growth factor; PF, platelet factor; VWF, von Willebrand factor.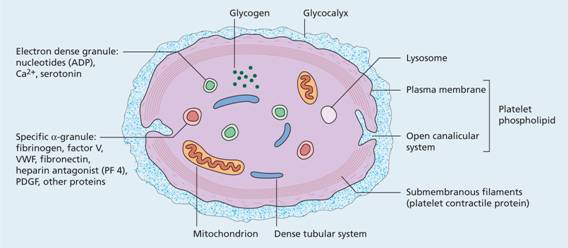
Figure 24.5 Platelet adhesion. The binding of glycoprotein (GP) Ib (which consists of four proteins: GPIb α, GPIb β, GPIX, GPV) to von Willebrand factor leads to adhesion to the subendothelium and also exposes the GPIIb/IIIa (αIIbβ3 integrin) binding sites to fibrinogen and von Willebrand factor leading to platelet aggregation. The GPIa site permits direct adhesion to collagen and also explores the GPIIb/IIIa binding site.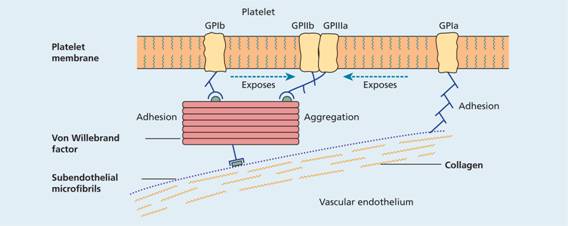
Figure 24.6 The synthesis of prostacyclin and thromboxane A2. The opposing effects of these agents are mediated by changes in the concentration of cyclic adenosine monophosphate (cAMP) in platelets via stimulation or inhibition of the enzyme adenylate cyclase. cAMP controls the concentration of free calcium ions in the platelet which are important in the processes that cause adhesion and aggregation. High levels of cAMP lead to low free calcium ion concentrations and prevent aggregation and adhesion. ATP, adenosine triphosphate; Ca, calcium; PG, prostaglandin (G2 and H2).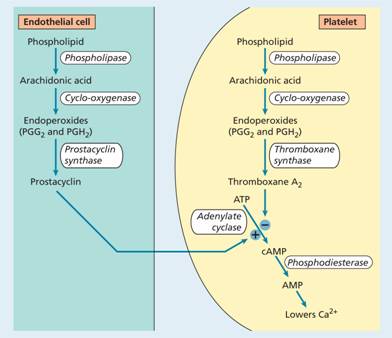
The plasma membrane invaginates into the platelet interior to form an open membrane (canalicular) system which provides a large reactive surface to which the plasma coagulation proteins may be selectively absorbed. The membrane phospholipids (previously known as platelet factor 3) are of particular importance in the conversion of coagulation factor X to Xa and prothrombin (factor II) to thrombin (factor IIa) (Fig. 24.7).
Figure 24.7 The pathway of blood coagulation initiated by tissue factor (TF) on the cell surface. When plasma comes into contact with TF, factor VII binds to TF. The complex of TF and activated VII (VIIa) activates X and IX. TF pathway inhibitor (TFPI) is an important inhibitor of TF/VIIa. The VIIIa–IXa complex greatly amplifies Xa production from X. The generation of thrombin from prothrombin by the action of Xa–Va complex leads to fibrin formation. Thrombin also activates XI (dashed line), V and XIII. Thrombin cleaves VIII from its carrier von Willebrand factor (VWF), greatly increasing the formation of VIIIa–IXa and hence of Xa–Va. Pale green, serine proteases; yellow, cofactors.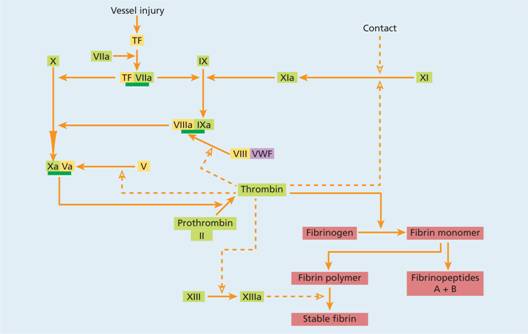
The platelet contains three types of storage granules: dense, α and lysosomes (Fig. 24.4). The more frequent specific α granules contain clotting factors, VWF, platelet-derived growth factor (PDGF) and other proteins. Dense granules are less common and contain adenosine diphosphate (ADP), adenosine triphosphate (ATP), serotonin and calcium. Lysosomes contain hydrolytic enzymes. Platelets are also rich in signalling and cytoskeletal proteins which support the rapid switch from quiescent to activation that follows vessel damage. During the release reaction described below, the contents of the granules are discharged into the open canalicular system.
Platelet antigens
Several platelet surface proteins have been found to be important antigens in platelet-specific autoimmunity and they have been termed human platelet antigens (HPA). In most cases, two different alleles exist, termed a or b alleles (e.g. HPA-1a). Platelets also express ABO and human leucocyte antigen (HLA) class I but not class II antigens.
Platelet function
The main function of platelets is the formation of mechanical plugs during the normal haemostatic response to vascular injury. In the absence of platelets, spontaneous leakage of blood through small vessels may occur. Platelet function falls into three: adhesion, aggregation and release reactions. There is also amplification. The immobilization of platelets at the sites of vascular injury requires specific platelet–vessel wall (adhesion) and platelet–platelet (aggregation) interactions, both partly mediated through VWF which is discussed next.
Von Willebrand factor
VWF is involved in shear dependent platelet adhesion to the vessel wall (see p. 318) and to other platelets (aggregation) (Fig. 24.5). It also carries factor VIII. It is a large cysteine-rich glycoprotein, with multimers made up on average of 2–50 dimeric subunits, with a molecular weight (MW) range of 0.8–20 × 106. VWF is encoded by a gene on chromosome 12 and is synthesized both in endothelial cells and megakaryocytes, and stored in Weibel–Palade bodies and platelet α granules, respectively.
Plasma VWF is almost entirely derived from endothelial cells, with two distinct pathways of secretion. The majority is continuously secreted and a minority is stored in Weibel–Palade bodies. The stored VWF can raise the plasma levels when released under the influence of several secretagogues, such as stress, exercise, adrenaline and infusion of desmopressin (1-diamino-8-D-arginine vasopressin; DDAVP). The VWF released from Weibel–Palade bodies is in the form of large and ultra large multimers, the most adhesive and reactive form of VWF. They are in turn cleaved in plasma to smaller multimers and monomeric VWF by the specific plasma metalloprotease, ADAMTS13 (see Fig. 25.7).
Platelet aggregation
This is characterized by cross-linking of platelets through active GPIIb/IIIa receptors with fibrinogen bridges. A resting platelet has about 50–80 000 GPIIb/IIIa receptors, which do not bind fibrinogen, VWF or other ligands. Stimulation of a platelet leads to an increase in GPIIb/IIIa molecules, enabling platelet cross-linking with fibrinogen bridges.
Platelet release reaction and amplification
Primary activation by various agonists induces intracellular signalling, leading to the release of α granule contents. These have an important role in platelet aggregate formation and stabilization and, in addition, the ADP released from dense granules has a major positive feedback role in promoting platelet activation.
Thromboxane A 2 (TXA2) is the second of the two major platelet positive feedback loops important in secondary amplification of platelet activation to form a stable platelet aggregate. It is formed de novo upon activation of cytosolic phospholipase A2 (PLA2) (Fig. 24.6). TXA2 is a labile substance and lowers platelet cyclic adenosine monophosphate (cAMP) levels and initiates the release reaction (Fig. 24.6). TXA2 not only potentiates platelet aggregation, but also has powerful vasoconstrictive activity. The release reaction is inhibited by substances that increase the level of platelet cAMP. One such substance is prostacyclin (PGI2) which is synthesized by vascular endothelial cells. It is a potent inhibitor of platelet aggregation and prevents their deposition on normal vascular endothelium.
Platelet procoagulant activity
After platelet aggregation and release, the exposed membrane phospholipid (platelet factor 3) is available for two reactions in the coagulation cascade. Both phospholipid-mediated reactions are calcium-ion dependent. The first (tenase) involves factors IXa, VIIIa and X in the formation of factor Xa (Fig. 24.7). The second (prothrombinase) results in the formation of thrombin from the interaction of factors Xa, Va and prothrombin (II). The phospholipid surface forms an ideal template for the crucial concentration and orientation of these proteins.
Growth factor
PDGF found in the α granules of platelets stimulates vascular smooth muscle cells to multiply and this may hasten vascular healing following injury.
Natural inhibitors of platelet function
Nitric oxide (NO) is constitutively released from endothelial cells (Fig. 24.8
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


