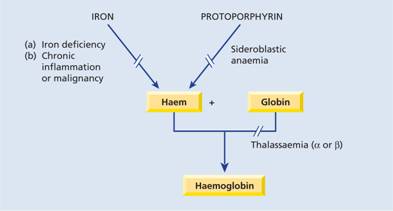
Nutritional and metabolic aspects of iron
Iron is one of the most common elements in the Earth’s crust, yet iron deficiency is the most common cause of anaemia, affecting about 500 million people worldwide. This is because the body has a limited ability to absorb iron and excess loss of iron as a result of haemorrhage is frequent. Also, in many developing countries, dietary intake is inadequate from childhood.
Body iron distribution and transport
The transport and storage of iron is largely mediated by three proteins: transferrin, transferrin receptor 1 (TfR1) and ferritin.
Transferrin can contain up to two atoms of iron. It delivers iron to tissues that have transferrin receptors, especially erythroblasts in the bone marrow which incorporate the iron into haemoglobin (Fig. 3.2). The transferrin is then reutilized. At the end of their life, red cells are broken down in the macrophages of the reticuloendothelial system and the iron is released from haemoglobin, enters the plasma and provides most of the iron on transferrin. Only a small proportion of plasma transferrin iron comes from dietary iron, absorbed through the duodenum and jejunum.
Figure 3.2 Daily iron cycle. Most of the iron in the body is contained in circulating haemoglobin (see Table 3.1) and is reutilized for haemoglobin synthesis after the red cells die. Iron is transferred from macrophages to plasma transferrin and so to bone marrow erythroblasts. Iron absorption is normally just sufficient to make up for iron loss. The dashed line indicates ineffective erythropoiesis.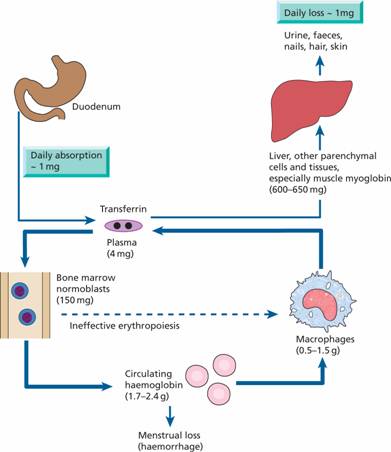
Some iron is stored in the macrophages as ferritin and haemosiderin, the amount varying widely according to overall body iron status. Ferritin is a water-soluble protein–iron complex of molecular weight 465000. It is made up of an outer protein shell, apoferritin, consisting of 22 subunits and an iron–phosphate–hydroxide core. It contains up to 20% of its weight as iron and is not visible by light microscopy. Each molecule of apoferritin may bind up to 4000–5000 atoms of iron.
Haemosiderin is an insoluble protein–iron complex of varying composition containing approximately 37% iron by weight. It is derived from partial lysosomal digestion of aggregates of ferritin molecules and is visible in macrophages and other cells by light microscopy after staining by Perls’ (Prussian blue) reaction. Iron in ferritin and haemosiderin is in the ferric form. It is mobilized after reduction to the ferrous form, vitamin C being involved. A copper-containing enzyme, caeruloplasmin, catalyses oxidation of the iron to the ferric form for binding to plasma transferrin.
Iron is also present in muscle as myoglobin and in most cells of the body in iron-containing enzymes (e.g. cytochromes, succinic dehydrogenase, catalase) (Table 3.1). This tissue iron is less likely to become depleted than haemosiderin, ferritin and haemoglobin in states of iron deficiency, but some reduction of haem-containing enzymes may occur.
Table 3.1 The distribution of body iron.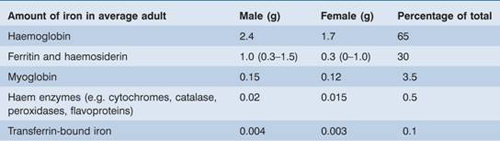
Regulation of ferritin and transferrin receptor 1 synthesis
The levels of ferritin, TfR1, δ-aminolevulinic acid synthase (ALA-S) and divalent metal transporter 1 (DMT-1) are linked to iron status so that iron overload causes a rise in tissue ferritin and a fall in TfR1 and DMT-1 whereas in iron deficiency ferritin and ALA-S are low and TfR1 increased. This linkage arises through the binding of an iron regulatory protein (IRP) to iron response elements (IREs) on the ferritin, TfR1, ALA-S and DMT-1 mRNA molecules. Iron deficiency increases the ability of IRP to bind to the IREs whereas iron overload reduces the binding. The site of IRP binding to IREs, whether upstream (5′) or downstream (3′) from the coding gene, determines whether the amount of mRNA and so protein produced is increased or decreased (Fig. 3.3). Upstream binding reduces translation whereas downstream binding stabilizes the mRNA, increasing translation and so protein synthesis.
Figure 3.3 Regulation of transferrin receptor 1 (TfR1), divalent metal transporter 1 (DMT-1) and ferritin expression by iron regulatory protein (IRP) sensing of intracellular iron levels. IRPs are able to bind to stem-loop structures called iron response elements (IREs). IRP binding to the IRE within the 3′ untranslated region of TfR1 and DMT-1 leads to stabilization of the mRNA and increased protein synthesis, whereas IRP binding to the IRE within the 5′ untranslated region of ferritin and δ-aminolevulinic acid synthase (ALA-S) mRNA reduces translation. IRPs can exist in two states: at times of high iron levels the IRP binds iron and exhibits a reduced affinity for the IREs whereas when iron levels are low the binding of IRPs to IREs is increased. In this way synthesis of TfR, ALA-S, DMT-1 and ferritin is coordinated to physiological requirements. IRP, iron regulatory protein.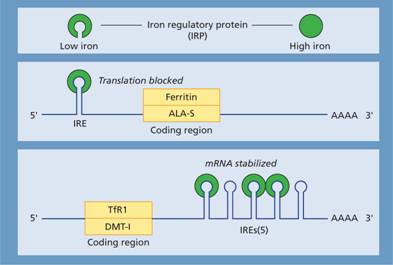
When plasma iron is raised and transferrin is saturated the amount of iron transferred to parenchymal cells (e.g. those of the liver, endocrine organs, pancreas and heart) is increased and this is the basis of the pathological changes associated with iron loading conditions. There may also be free iron in plasma which is toxic to different organs.
Hepcidin
Hepcidin is a 25amino acid polypeptide produced by liver cells. It is the major hormonal regulator of iron homeostasis (Fig. 3.4). It inhibits iron release from macrophages and intestinal epithelial cells by its interaction with the transmembrane iron exporter ferroportin, accelerating degradation of ferroportin mRNA. Raised hepcidin levels therefore reduce iron absorption and iron release from macrophages.
Figure 3.4 The role of hepcidin in the regulation of iron absorption and iron release from macrophages. BMP, bone morphogenetic protein; FPN, ferroportin; HJV, hemojuvelin; IL-6, interleukin 6; TF, transferrin, transcription factor Smad4 which stimulates hepcidin synthesis.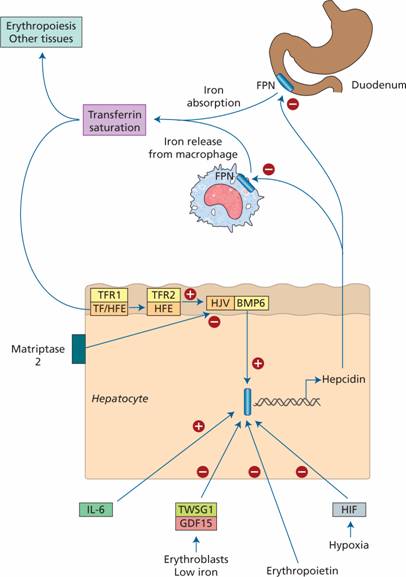
Control of hepcidin expression
Membrane bound hemojuvelin (HJV) is a co-receptor with bone morphogenetic protein (BMP) which stimulates hepcidin expression. A complex between HFE and transferrin receptor 2 (TfR2) promotes HJV binding to BMP. The amount of HFE–TfR2 complex is determined by the degree of iron saturation of transferrin as follows. Diferric transferrin competes with TfR1 for binding to HFE. The more diferric transferrin, the less TfR1 is bound to HFE and more HFE is available to bind to TfR2 with consequently increased hepcidin synthesis. Low concentrations of diferric transferrin, as in iron deficiency, allow HFE binding to TfR1, reducing the amount of HFE able to bind TfR2 and thus reducing hepcidin secretion.
Matriptase 2 digests membrane-bound HJV. In iron deficiency, increased matriptase activity therefore results in decreased hepcidin synthesis. Erythroblasts secrete two proteins, GDF 15 and TWSG1, which suppress hepcidin secretion. In conditions with increased numbers of early erythroblasts in the marrow (e.g. conditions of ineffective erythropoiesis such as thalassaemia major), iron absorption is increased because of suppression of hepcidin secretion by these two proteins. Hypoxia also suppresses hepcidin synthesis whereas in inflammation interleukin 6 (IL-6) and other cytokines increase hepcidin synthesis (Fig. 3.4).
Dietary iron
Iron is present in food as ferric hydroxides, ferric–protein and haem–protein complexes. Both the iron content and the proportion of iron absorbed differ from food to food; in general, meat – in particular liver – is a better source than vegetables, eggs or dairy foods. The average Western diet contains 10–15 mg iron daily from which only 5–10% is normally absorbed. The proportion can be increased to 20–30% in iron deficiency or pregnancy (Table 3.2) but even in these situations most dietary iron remains unabsorbed.
Table 3.2 Iron absorption.
| Factors favouring absorption | Factors reducing absorption |
| Haem iron | Inorganic iron |
| Ferrous form (Fe2+) | Ferric form (Fe3+) |
| Acids (HCl, vitamin C) | Alkalis – antacids, pancreatic secretions |
| Solubilizing agents (e.g. sugars, amino acids) | Precipitating agents – phytates, phosphates, tea |
| Reduced serum hepcidin, e.g. iron deficiency | Increased serum hepcidin, e.g. iron excess |
| Ineffective erythropoiesis | Decreased erythropoiesis |
| Pregnancy | Inflammation |
| Hereditary haemochromatosis | |
| Increased expression of DMT-1 in duodenal enterocytes | Decreased expression of DMT-1 in duodenal enterocytes |
Iron absorption
Organic dietary iron is partly absorbed as haem and partly broken down in the gut to inorganic iron. Absorption occurs through the duodenum. Haem is absorbed through a receptor, yet to be identified, on the apical membrane of the duodenal enterocyte. Haem is then digested to release iron. Inorganic iron absorption is favoured by factors such as acid and reducing agents that keep iron in the gut lumen in the Fe2+ rather than the Fe3+ state (Table 3.2). The protein DMT-1 is involved in transfer of iron from the lumen of the gut across the enterocyte microvilli (Fig. 3.5). Ferroportin at the basolateral surface controls exit of iron from the cell into portal plasma.The amount of iron absorbed is regulated according to the body’s needs by changing the levels of DMT-1 and ferroportin. For DMT-1 this occurs by the same mechanism (IRP/IRE binding) by which transferrin receptor is increased in iron deficiency (Fig. 3.3). Hepcidin is a major regulator by affecting ferroportin concentration. Low hepcidin levels in iron deficiency allow increased ferroportin levels and so more iron to enter portal plasma.
Figure 3.5 The regulation of iron absorption. Dietary ferric (Fe3+) iron is reduced to Fe2+ and its entry to the enterocyte is through the divalent cation binder DMT-1. Its export into portal plasma is controlled by ferroportin. It is oxidized before binding to transferrin in plasma. Haem is absorbed after binding to its receptor protein.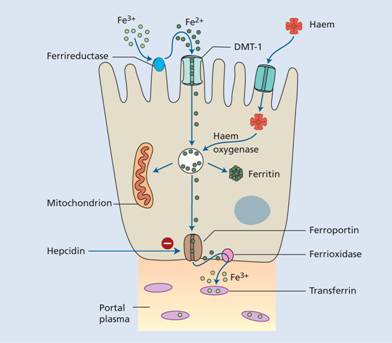
Ferrireductase present at the apical surface converts iron from the Fe3+ to Fe2+ state and another enzyme, hephaestin (ferrioxidase) (which contains copper), converts Fe2+ to Fe3+ at the basal surface prior to binding to transferrin.
Iron requirements
The amount of iron required each day to compensate for losses from the body and for growth varies with age and sex; it is highest in pregnancy, adolescent and menstruating females (Table 3.3). Therefore these groups are particularly likely to develop iron deficiency if there is additional iron loss or prolonged reduced intake.
Table 3.3 Estimated daily iron requirements. Units are mg/day.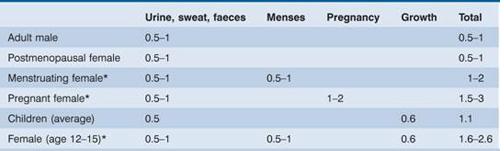
* These groups are more likely to develop iron deficiency.
Clinical features
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


