Pancytopenia
Pancytopenia is a reduction in the blood count of all the major cell lines – red cells, white cells and platelets. It has several causes (Table 22.1) which can be broadly divided into decreased bone marrow production or increased peripheral destruction.
Table 22.1 Causes of pancytopenia.
| Decreased bone marrow function |
| Aplasia |
| Acute leukaemia, myelodysplasia, myeloma |
| Infiltration with lymphoma, solid tumours, tuberculosis |
| Megaloblastic anaemia |
| Paroxysmal nocturnal haemoglobinuria |
| Myelofibrosis |
| Haemophagocytic syndrome |
| Increased peripheral destruction |
| Splenomegaly |
Aplastic (hypoplastic) anaemia is defined as pancytopenia resulting from aplasia of the bone marrow. It is classified into primary (congenital or acquired) or secondary types (Table 22.2).
Table 22.2 Causes of aplastic anaemia.
| Primary | Secondary |
| Congenital (Fanconi and non-Fanconi types) | Ionizing radiation: Accidental exposure (radiotherapy, radioactive isotopes, nuclear power stations) |
| Idiopathic acquired | Chemicals: Benzene, organophosphates and other organic solvents, DDT and other pesticides, organochlorines, recreational drugs (ecstasy) |
| Drugs: Those that regularly cause marrow depression (e.g. busulfan, melphalan, cyclophosphamide, anthracyclines, nitrosoureas). | |
| Those that occasionally or rarely cause marrow depression (e.g. chloramphenicol, sulphonamides, gold, anti-inflammatory, antithyroid, psychotropic, anticonvulsant/antidepressant drugs) | |
| Viruses: Viral hepatitis (non-A, non-B, non-C, in most cases), EBV |
EBV, Epstein–Barr virus.
Pathogenesis
The underlying defect in all cases appears to be a substantial reduction in the number of haemopoietic pluripotential stem cells, and a fault in the remaining stem cells or an immune reaction against them, which makes them unable to divide and differentiate sufficiently to populate the bone marrow (Fig. 22.1). A primary fault in the marrow microenvironment has also been suggested but the success of stem cell transplantation (SCT) shows this can only be a rare cause because normal donor stem cells are usually able to thrive in the recipient’s marrow cavity.
Figure 22.1 Aplastic anaemia: low power views of bone marrow show severe reduction of haemopoietic cells with an increase in fat spaces.(a) Aspirated fragment.(b) Trephine biopsy. 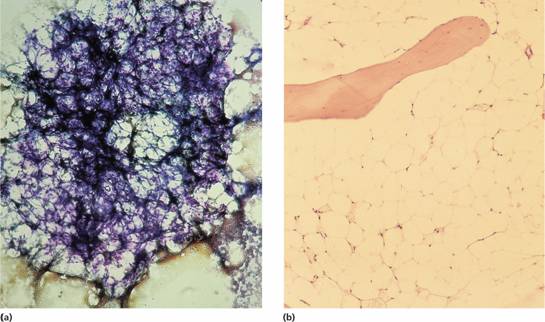
Congenital
The Fanconi type has an autosomal recessive pattern of inheritance and is often associated with growth retardation and congenital defects of the skeleton (e.g. microcephaly, absent radii or thumbs), of the renal tract (e.g. pelvic or horseshoe kidney) (Fig. 22.2) or skin (areas of hyper- and hypopigmentation); sometimes there is mental retardation. The syndrome is genetically heterogeneous with 13 different genes involved, A, B, C, D1, D2, E, F, G, I, J, L, M and N in different families. The encoded proteins cooperate in a common cellular pathway which results in ubiquitination of FANCD2, which protects cells against genetic damage.
Figure 22.2 (a) X-rays showing absent thumbs in a patient with Fanconi’s anaemia (FA).(b) Intravenous pyelogram in a patient with FA showing a normal right kidney but a left kidney abnormally placed in the pelvis.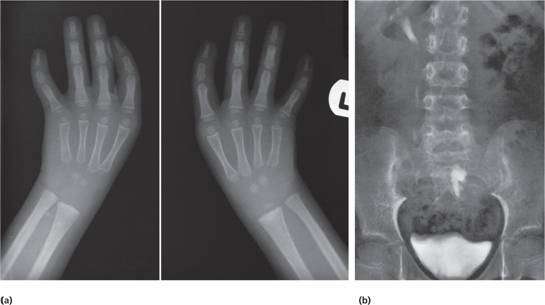
Cells from patients with Fanconi’s anaemia (FA) show an abnormally high frequency of spontaneous chromosomal breakage and the diagnostic test is elevated breakage after incubation of peripheral blood lymphocytes with the DNA cross-linking agent diepoxybutane (DEB test).
The usual age of presentation of FA is 5–10 years. Approximately 10% of patients develop acute myeloid leukaemia. Treatment is usually with androgens and/or SCT. The blood count usually improves with androgens but side-effects, especially in children, are distressing (virilization and liver abnormalities); remission rarely lasts more than 2 years. SCT may cure the patient. Because of the sensitivity of the patient’s cells to DNA damage, conditioning regimens are mild.
Dyskeratosis congenita (DC) is a rare sex-linked disorder with nail and skin atrophy, aplastic anaemia and a high risk of cancer. It is associated with mutations in theDKC1 (dyskerin) orTERC (telomerase reverse transcriptase RNA template) genes which are both involved in the maintenance of telomere length.
Other inherited bone marrow failure syndromes include Diamond–Blackfan anaemia (DBA), (see p. 294), Shwachman–Diamond syndrome (SDS), (see p. 295), severe congenital neutropenia (see p. 119), amegakaryocytic thrombocytopenia (see p. 333) and thrombocytopenia with absent radii (see p. 333). In DC, DBA and SDS there are defects in ribosomal biosynthesis and function (Fig. 22.3).
Figure 22.3 Schematic showing scheme of rRNA processing in human cells and the points at which this is possibly disrupted in the different bone marrow failure syndromes. The ribosomal RNAs (rRNAs) are transcribed by RNA polymerase I as a single precursor transcript (45S rRNA). The 45S rRNA is then processed to 18S, 5.8S and 28S rRNAs. The 18S is a component of the 40S ribosomal subunit. The 5.8S and 28S together with 5S (synthesized independently) are components of the 60S ribosomal subunit. The 40S and 60S subunits are assembled to form the 80S ribosomes. The processing steps affected in Diamond – Blackfan anaemia (due to heterozygous mutations inRPS19, RPS17, RPS24, RPS7, RPL35a, RPL5, RPL11), 5q-syndrome (haploinsufficiency ofRPS14) and Shwachman–Diamond syndrome (biallelic mutations inSBDS) are indicated by the different coloured stars. DBA, Diamond Blackfan anaemia; SDS, Shwachman–Diamond syndrome; S, sedimentation coefficient.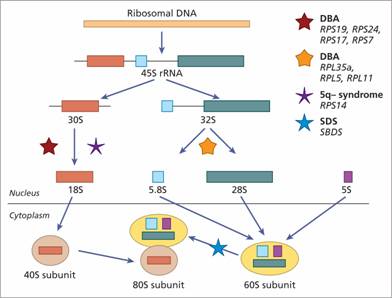
Idiopathic acquired
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


