Incidence and pathogenesis
The incidence of ALL is highest at 3–7 years with 75% of cases occurring before the age of 6. There is a secondary rise after the age of 40 years. Eighty-five per cent of cases are of B-cell lineage and have an equal sex incidence; there is a male predominance for the 15% of T-cell ALL (T-ALL).
The pathogenesis is varied. Certain germline polymorphism in a group of genes mainly involved in B-cell development (e.g. IKZF1) are more frequent in patients with B-cell ALL (B-ALL) than controls. Interestingly, IKZF1 is also deleted in the leukaemic cells in 30% of high risk B-ALL and 95% of ALL BCR-ABL1 positive cases. In a proportion of cases the first event occurs in the fetus in utero, with a secondary event possibly precipitated by infection in childhood (see Fig. 11.3). The first event is a translocation (e.g. t (12; 21)) or point mutation. The second event involves genome-wide copy number alterations, some of which encode for functions relevant to leukaemogenesis. This is discussed further in Chapter 11. In other cases, the disease seems to arise as a postnatal mutation in an early lymphoid progenitor cell.
Acute lymphoblastic leukaemia, B cell or T cell, is subclassified by WHO (2008) according to the underlying genetic defect (Table 17.1). Within B-ALL there are several specific genetic subtypes such as those with the t (9; 22) or t (12; 21) translocations, rearrangements of the MLL gene or alteration in chromosome number (diploidy) (Table 17.1). The subtype is an important guide to the optimal treatment protocol and to prognosis. In T-ALL an abnormal karyotype is found in 50–70% of cases and the NOTCH signalling pathway is activated in most cases (see below).
Table 17.1 Classification of acute lymphoblastic leukaemia (ALL) according to the World Health Organization (WHO) classification (modified).
| Precursor lymphoid neoplasms |
| B lymphoblastic leukaemia/lymphoma |
| B lymphoblastic leukaemia/lymphoma, NOS |
| B lymphoblastic leukaemia/lymphoma with recurrent genetic abnormalities |
| B lymphoblastic leukaemia/lymphoma with t (9; 22)(q34; q11.2); BCR-ABL1 |
| B lymphoblastic leukaemia/lymphoma with t (v; 11q23); MLL rearranged |
| B lymphoblastic leukaemia/lymphoma with t (12; 21) (p13; q22); TEL-AML1 (ETV6-RUNX1) |
| B lymphoblastic leukaemia/lymphoma with hyperdiploidy |
| B lymphoblastic leukaemia/lymphoma with hypodiploidy (hypodiploid ALL) |
| T lymphoblastic leukaemia/lymphoma |
NOS, not otherwise specified.
Clinical features are a result of the following.
Bone marrow failure
- Anaemia (pallor, lethargy and dyspnoea);
- Neutropenia (fever, malaise, features of mouth, throat, skin, respiratory, perianal or other infections);
- Thrombocytopenia (spontaneous bruises, purpura, bleeding gums and menorrhagia).
Organ infiltration
Tender bones, lymphadenopathy (Fig. 17.1a), moderate splenomegaly, hepatomegaly and meningeal syndrome (headache, nausea and vomiting, blurring of vision and diplopia). Fundal examination may reveal papilloedema and sometimes haemorrhage. Many patients have a fever which usually resolves after starting chemotherapy. Less common manifestations include testicular swelling (Fig. 17.1b) or signs of mediastinal compression in T-ALL (Fig. 17.2).
Figure 17.1 Acute lymphoblastic leukaemia. (a) Marked cervical lymphadenopathy in a boy. (b) Testicular swelling and erythema on the left-hand side of the scrotum caused by testicular infiltration. (Courtesy of Professor J.M. Chessels.)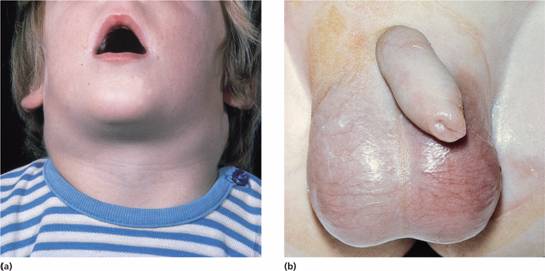
Figure 17.2 Chest X-ray of a boy aged 16 years with acute lymphoblastic leukaemia (T-ALL). (a) There is a large mediastinal mass caused by thymic enlargement at presentation. (b) After 1 week of therapy with prednisolone, vincristine and daunorubicin the mass has resolved.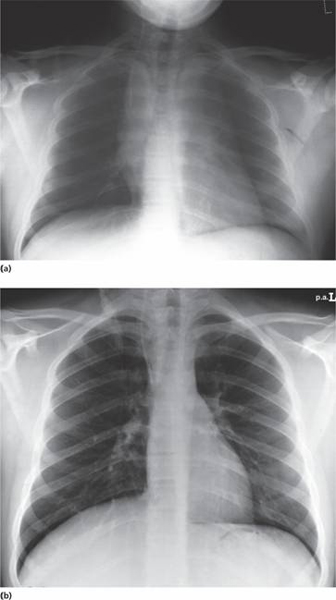
If lymph node or solid extranodal masses predominate with <20% blasts in the marrow the disease is called lymphoblastic lymphoma but is treated as ALL.
Haematological investigations reveal a normochromic normocytic anaemia with thrombocytopenia in most cases. The total white cell count may be decreased, normal or increased to 200 × 109/L or more. The blood film typically shows a variable numbers of blast cells. The bone marrow is hypercellular with >20% leukaemic blasts. The blast cells are characterized by morphology (Fig. 17.3), cytochemisty (Table 17.2), immunological tests (Table 17.3) and cytogenetic analysis (Table 17.1). Identification of the immunoglobulin or T-cell receptor (TCR) gene rearrangement, (aberrant) immunophenotype and molecular genetics of the tumour cells is important to determine treatment and to detect minimal residual disease (MRD) during follow-up.
Figure 17.3 Morphology, cytochemistry and immunophenotyping of acute lymphoblastic leukaemia (ALL). (a) Lymphoblasts show scanty cytoplasm without granules. (b) Lymphoblasts are large and heterogeneous with abundant cytoplasm. (c) Lymphoblasts are deeply basophilic with cytoplasmic vacuolation. (d) Indirect immunofluorescence reveals nuclear terminal deoxynucleotidyl transferase (TdT) (green) and membrane CD10 (orange). (Courtesy of Professor G. Janossy.)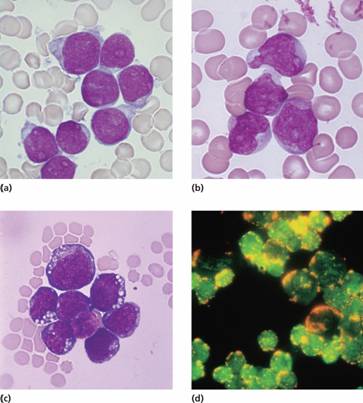
Table 17.2 Specialized tests for acute lymphoblastic leukaemia (ALL).
| Cytochemistry | |
| Myeloperoxidase | − |
| Sudan black | − |
| Non-specific esterase | − |
| Periodic acid–Schiff | + (coarse block positivity in ALL) |
| Acid phosphatase | + in T-ALL (Golgi staining) |
| Immunoglobulin and TCR genes | B-ALL: clonal rearrangement of immunoglobulin genes |
| T-ALL: clonal rearrangement of TCR genes | |
| Chromosomes and genetic analysis | (Table 17.1) |
| Immunological markers (flow cytometry) | (Table 17.3) |
B-ALL, B-cell acute lymphoblastic leukaemia; T-ALL, T-cell acute lymphoblastic leukaemia; TCR, T-cell receptor.
Table 17.3 Immunological markers for classification of acute lymphoblastic (ALL) leukaemia.
| ALL | ||
| Marker | B | T |
| B lineage | ||
| CD19 | + | − |
| cCD22 | + | − |
| cCD79a | + | − |
| CD10 | + or − | − |
| cIg | + (pre-B) | − |
| sIg | − | − |
| TdT | + | + |
| lineage | ||
| CD7 | − | + |
| cCD3 | − | + |
| CD2 | − | + |
| TdT | ||



