Myelodysplasia (myelodysplasticsyndromes)
This is a group of clonal disorders of haemopoietic stem cells characterized by increasing bone marrow failure in association with quantitative and qualitative abnormalities of cells in peripheral blood (Table 16.1). A hallmark of the disease is simultaneous proliferation and apoptosis of haemopoietic cells (ineffective haemopoiesis) leading to the paradox of a hypercellular bone marrow but pancytopenia in peripheral blood. There is a tendency to progress to acute myeloid leukaemia (AML), although death often occurs before this develops.
Table 16.1 The World Health Organization (2008) classification of myelodysplasia.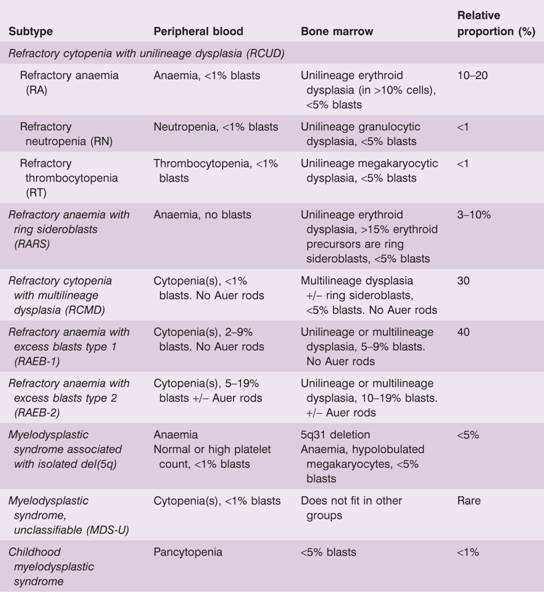
In most cases, the disease is primary but in a significant proportion of patients it is secondary to chemotherapy and/or radiotherapy that has been given for treatment of another malignancy. This latter type is termed therap-related MDS (t-MDS) and is now classified with therapy-related AML.
The pathogenesis of myelodysplastic syndromes (MDS) is unclear but is presumed to start following genetic damage to a multipotent haemopoietic progenitor cell. The immune system may have a minor role in suppressing bone marrow function and immunosuppression is sometimes used in treatment (see below).
Classification
The myelodysplastic syndromes are classified on the basis of the blood count, their morphological appearance, the number of blast cells in blood or bone marrow and cytogenetics (Table 16.1). Although the classification appears complex, the principles are as follow:
- Dysplasia may be present solely in a single lineage – red cells (refractory anaemia), neutrophils or platelets – or present in two or more myeloid lineages (refractory cytopenia with multilineage dysplasia; RCMD).
- Erythroid dysplasia can also be associated with ring sideroblasts to define a unique subtype. The definition of a pathological ring sideroblast is an erythroid precursor with five or more iron granules encircling at least one-third of the nucleus. A rare condition, refractory anaemia with ring sideroblasts and thrombocytosis (platelets >450 × 109/L) in which the JAKV617F mutation is often present, is classified with the myelodysplastic–myeloproliferative diseases (Table 16.4).
- If the blast cell count is increased in blood or bone marrow, the diagnosis is made of refractory anaemia with excess blasts and these subtypes have a poor prognosis.
- 5q-Syndrome receives its own classification. The gene that is deleted is RPS14, encoding a ribosomal protein (see Fig. 22.3). It is more common in women and typically there is anaemia thrombocytosis in 50% of cases. It has a particularly good prognosis (Table 16.3).
Table 16.2 Cytogenetic abnormalities inmyelodysplastic syndromes.
| 1 Chromosome deletion or loss (e.g.del 5q, monosomy 5, del 7q. monosomy 7, del 11q) |
| 2 Chromosome gain (e.g. trisomy 8, trisomy 11) |
| 3 Chromosome rearrangement (e.g. t3q26, t(1; 7), t11q23) |
| 4 Complex karyotypes: three or more abnormalities |
Table 16.3 Classification of risk group in myelodysplastic syndromes (MDS).
| Lower risk MDS | Higher risk MDS |
| Survival of 3–10 years | Survival <1.5 years |
| Low rate of transformation to AML | High rate of AML transformation |
| RA, RARS | RAEB |
| RCUD, RCMD | |
| MDS-U, MDS del(5q) | |
| IPSS low | IPSS high |
AML, acute myeloid leukaemia; IPSS, International Prognostic Scoring System; RA, refractory anaemia; RAEB, refractory anaemia with excess blasts; RARS, refractory anaemia with ring sideroblasts; RCUD, refractory cytopenia with unilineage dysplasia; RCMD, refractory cytopenia with multilineage dysplasia; MDS, myelodys-plastic syndrome; MDS-U, myelodysplastic syndrome, unclassifiable.
Table 16.4 Classification of myelodysplastic/myeloproliferative neoplasms.
| Diagnostic features | |
| Chronic myelomonocytic leukaemia | Monocytosis >1 × 109/L |
| Atypical chronic myeloid leukaemia | WBC >13 × 109/L BCR-ABL1 absent |
| BCR-ABL1 negative | |
| Juvenile myelomonocytic leukaemia | |
| Myelodysplastic/myeloproliferative neoplasm, unclassifiable | |
| Refractory anaemia with ring sideroblasts associated with marked thrombocytosis | Platelet count >450 × 109/L Large atypical megakaryocytes |
Clinical features
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


