Table 15.1 Myeloproliferative diseases and other myeloid neoplasms associated with point mutation or rearrangement of tyrosine kinase genes.
| Disease | Tyrosine kinase gene mutated |
| Chronic myeloid leukaemia | ABL1 |
| Polycythaemia vera | JAK2 V617F; JAK2 exon12 |
| Primary myelofibrosis | JAK2 V617F; MPL W151L/K |
| Essential thrombocythaemia | JAK2 V617F; MPL W151L/K |
| Mastocytosis | KIT D816V |
| Myeloid neoplasm with eosinophilia | PDGFRA, PDGFRB, FGFR1 |
Figure 15.2 The role of JAK2 mutation in the generation of myeloproliferative diseases. (a) (i) Most haemopoietic growth factor receptors do not have intrinsic kinase activity but associate with a protein kinase such as JAK2 in the cytoplasm. (ii) When the receptor binds a growth factor the cytoplasmic domains move closer together and the JAK2 molecules can activate each other by phosphorylation. (iii) The V617F JAK2 mutation allows the JAK protein to become activated even when no growth factor is bound. (b) DNA sequencing shows homozygous G → T mutation in JAK2 in granulocytes but not in T lymphocytes (left-hand panel) and heterozygous mutation in right-hand panel. (After Kralovic R. et al.N Engl J Med 2005, 352, 1779–90.) (c) JAK2 activation leads to cell survival and proliferation through activation of three major pathways; the STAT transcription factors, the PI3K pathway acting through Akt and Ras activation which subsequently activate ERK and MAPK. The net result is production of a diverse range of proteins that promote cell survival and proliferation. (d) A model for the development of myeloproliferative disease following JAK2 mutation. The primary event appears to predispose to an acquired heterozygous mutation of JAK2 (V617F). This leads to a survival advantage. In some patients, a mitotic recombination event leads to a homozygous JAK2 mutation state.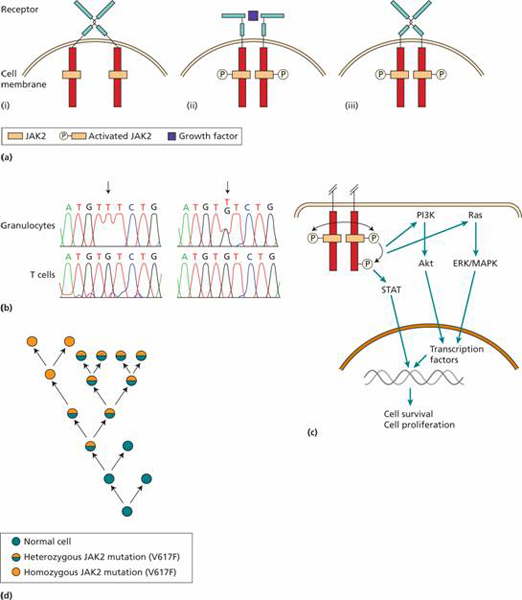
Polycythaemia is defined as an increase in the haemoglobin concentration above the upper limit of normal for the patient’s age and sex.
Classification of polycythaemia
Polycythaemia is classified according to its pathophysiology but the major subdivision is into absolute polycythaemia or erythrocytosis, in which the red cell mass (volume) is raised to greater than 125% of that expected for body mass and gender, and relative or pseudopolycythaemia in which the red cell volume is normal but the plasma volume is reduced. If the haematocrit is >0.60 then there will always be a raised red cell mass. Hb >18.5 g/dL or haematocrit >0.52 in men, and Hb >16.5 g/dL or haematocrit >0.48 in women, indicate that erythrocytosis is likely but isotope studies may be required (Table 15.2).
Table 15.2 Radiodilution methods for measuring red cell and plasma volume.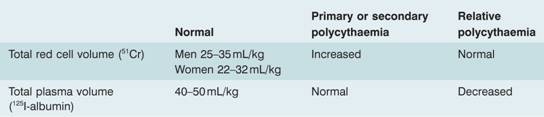
Once established, absolute polycythaemia can then be subdivided into primary polycythaemia (in which the erythroid progenitor cell shows an enhanced response to cytokines) or secondary polycythaemia (driven by factors outside the erythroid compartment) (Table 15.3).
Table 15.3 Causes of polycythaemia (erythrocytosis).
| Primary erythrocytosis |
| Congenital |
| Erythropoietin receptor mutations |
| Acquired |
| Polycythaemia vera |
| Secondary erythrocytosis |
| Congenital |
| Defects of the oxygen-sensing pathway |
| VHL gene mutation (Chuvash erythrocytosis) |
| PHD2 mutations |
| HIF-2α mutations |
| Other congenital defects |
| High oxygen-affinity haemoglobin |
| Acquired |
| Erythropoietin–mediated |
| Central hypoxia |
| Chronic lung disease |
| Right to left cardiopulmonary vascular shunts |
| Carbon monoxide poisoning |
| Smoking |
| Obstructive sleep apnoea |
| High altitude |
| Local hypoxia |
| Renal artery stenosis |
| End-stage renal disease |
| Hydronephrosis |
| Renal cysts (polycystic kidney disease) |
| Post-renal transplant erythrocytosis |
| Pathologic erythropoietin production |
| Tumours–cerebellar haemangioblastoma, meningioma, parathyroid tumours, hepatocellular carcinoma, renal cell cancer, phaeochromocytoma, uterine leiomyoma |
| Drug–associated |
| Erythropoietin administration |
| Androgen administration |
Primary polycythaemia (erythrocytosis)
Congenital
(See below.)
Acquired
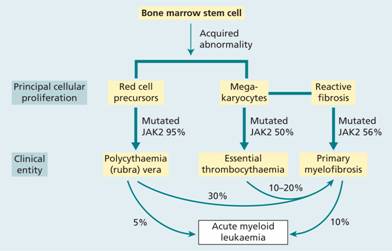
This is caused by the acquisition of mutations in the JAK2 gene leading to PV.
In PV, the increase in red cell volume is caused by a clonal malignancy of a marrow stem cell. The disease results from somatic mutation of a single haemopoietic stem cell which gives its progeny a proliferative advantage. The Val617Phe JAK2 mutation is present in haemopoietic cells in over 95% of patients and a mutation in exon 12 is seen in some of the remainder. Although the increase in red cells is the diagnostic finding, in many patients there is also an overproduction of granulocytes and platelets. Some families have an inherited predisposition to myeloproliferative disease and, interestingly, although affected individuals acquire JAK2 mutations in the marrow, these are not present in the germline.
Diagnosis
Making the diagnosis of PV in a patient who presents with polycythaemia can be difficult and two subsets are recognized based on the presence of the JAK2 mutation (Table 15.4).
Table 15.4 Criteria for diagnosis of polycythaemia vera. (From McMullin M.F. et al., (2007) B J Haem 138: 821.)
| JAK2-positive polycythaemia vera | |
| A1 | High haematocrit (> 0.52 in men, >0.48 in women) or raised red cell mass (> 25% above predicted) * |
| A2 | Mutation in JAK2 |
| Diagnosis requires both criteria to be present | |
| JAK2-negative polycythaemia vera | |
| A1 | Raised red cell mass (> 25% above predicted) or haematocrit >0.60 in men, >0.56 in women. |
| A2 | Absence of mutation in JAK2 |
| A3 | No cause of secondary erythrocytosis |
| A4 | Palpable splenomegaly |
| A5 | Presence of an acquired genetic abnormality (excluding BCR-ABL) in the haematopoietic cells |
| B1 | Thrombocytosis (platelet count >450×109/L) |
| B2 | Neutrophil leucocytosis (neutrophil count >10×109/L in non-smokers; >12.5×109/L in smokers) |
| B3 | Radiological evidence of splenomegaly |
| B4 | Endogenous erythroid colonies or low serum erythropoietin |
| Diagnosis requires A1 + A2 + A3 + either another A or two B criteria | |
* WHO (2008) uses haemoglobin >18.5 g/dL in men and 16.5 g/dL in women as a major criterion, in JAK2 + cases and hypercellular marrow as a minor criterion as well as criteria A2 and B4 above.
Clinical features
This is a disease of older subjects with an equal sex incidence. Clinical features are the result of hyperviscosity, hypervolaemia or hypermetabolism.
1 Headaches, dyspnoea, blurred vision and night sweats. Pruritus, characteristically after a hot bath, can be a severe problem.
2 Plethoric appearance: ruddy cyanosis (Fig. 15.3), conjunctival suffusion and retinal venous engorgement.
3 Splenomegaly in 75% of patients (Fig. 15.4).
4 Haemorrhage (e.g. gastrointestinal, uterine, cerebral) or thrombosis either arterial (e.g. cardiac, cerebral, peripheral) or venous (e.g. deep or superficial leg veins, cerebral, portal or hepatic veins) are frequent.
5 Hypertension in one-third of patients.
6 Gout (as a result of raised uric acid production; Fig. 15.5a).
Figure 15.3 Polycythaemia vera: facial plethora and conjunctival suffusion in a 63-year-old woman. Haemoglobin 18 g/dL; total red cell volume 45 mL/kg.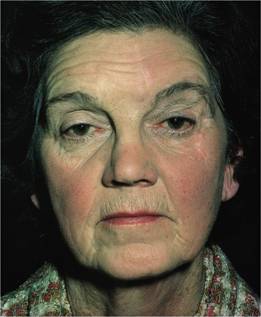
Figure 15.4 Splenomegaly: enlarged spleens in male patients with (a) polycythaemia vera and (b) myelofibrosis.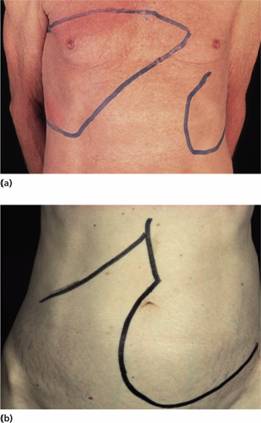
Figure 15.5 (a) The feet of a 72-year-old man with polycythaemia rubra vera. There is inflammation of the right metatarsophalangeal and other joints caused by uric acid deposits. (b)
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


