positive CML
Chronic myeloid (myelogenous) leukaemia BCR-ABL1 positive
Chronic myeloid leukaemia BCR-ABL1+ (CML) is a clonal disorder of a pluripotent stem cell. The disease accounts for around 15% of leukaemias and may occur at any age. The diagnosis of CML is rarely difficult and is assisted by the characteristic presence of the Philadelphia (Ph) chromosome. This results from the t(9; 22) (q34; q11) translocation between chromosomes 9 and 22 as a result of which part of the oncogene ABL1 is moved to the BCR gene on chromosome 22 (Fig. 14.1a) and part of chromosome 22 moves to chromosome 9. The abnormal chromosome 22 is the Ph chromosome. In the Ph translocation 5′ exons of BCR are fused to the 3′ exons of ABL1 (Fig. 14.1 b,c). The resulting chimeric BCR-ABL1 gene codes for a fusion protein of size 210 kDa (p210). This has tyrosine kinase activity in excess of the normal 145-kDa ABL1 product. The Ph translocation is also seen in a minority of cases of acute lymphoblastic leukaemia (ALL) and in some of these the breakpoint in BCR occurs in the same region as in CML. However, in other cases the breakpoint in BCR is further upstream, in the intron between the first and second exons, leaving only the first BCR exon intact. This chimeric BCR-ABL1 gene is expressed as a p190 protein which, like p210, has enhanced tyrosine kinase activity.
Figure 14.1 The Philadelphia chromosome. (a) There is translocation of part of the long arm of chromosome 22 to the long arm of chromosome 9 and reciprocal translocation of part of the long arm of chromosome 9 to chromosome 22 (the Philadelphia chromosome). This reciprocal translocation brings most of the ABL gene into the BCR region on chromosome 22 (and part of the BCR gene into juxtaposition with the remaining portion of ABL on chromosome 9). (b) The breakpoint in ABL is between exons 1 and 2. The breakpoint in BCR is at one of the two points in the major breakpoint cluster region (M-BCR) in chronic myeloid leukaemia (CML) or in some cases of Ph+acute lymphoblastic leukaemia (ALL). (c) This results in a 210-kDa fusion protein product derived from the BCR-ABL fusion gene. In other cases of Ph+ALL, the breakpoint in BCR is at a minor breakpoint cluster region (m-BCR) resulting in a smaller BCR-ABL fusion gene and a 190-kDa protein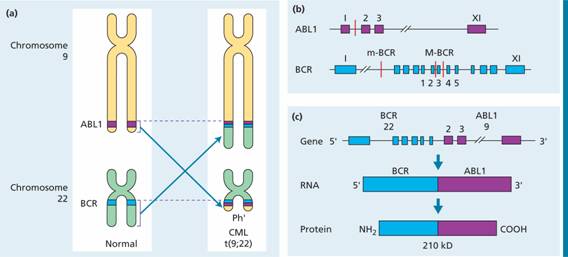
Figure 14.1 (Continued) (d) Karyotype showing the t(9; 22) (q34; q11) translocation. The Ph chromosome is arrowed. (e) Visualization of the Philadelphia chromosome on: (i) dividing (metaphase); and (ii) quiescent (interphase) cells by fluorescence in situ hybridization (FISH) analysis (ABL probe in red and BCR probe in green) with fusion signals (red/green) on the Ph and der(9) chromosomes. (Courtesy of Dr Ellie Nacheva)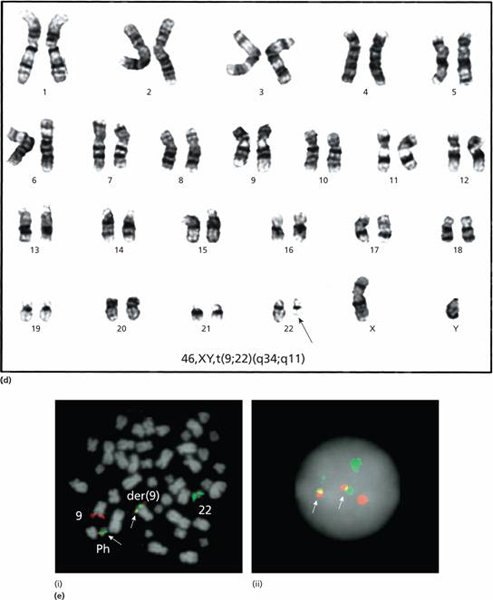
In most patients the Ph chromosome is seen by karyotypic examination of tumour cells (Fig. 14.1d) but in a few the Ph abnormality cannot be seen under the microscope but the same molecular rearrangement is detectable by more sensitive techniques: fluorescence in situ hybridization (FISH) (Fig. 14.1e) or polymerase chain reaction (PCR). Ph-negative BCR-ABL1 positive CML behaves clinically like Ph-positive CML. As the Ph chromosome is an acquired abnormality of haemopoietic stem cells it is found in cells of both the myeloid (granulocytic, erythroid and megakaryocytic) and lymphoid (B and T cell) lineages. Ph–, BCR-ABL1–chronic myeloid leukaemia is classified with the myelodysplastic/myeloproliferative syndromes (see Chapter 16).
Clinical features
This disease occurs in either sex (male: female ratio of 1.4: 1), most frequently between the ages of 40 and 60 years. However, it may occur in children and neonates, and in the very old. In most cases there are no predisposing factors but the incidence was increased in survivors of the atom bomb exposures in Japan. Its clinical features include the following:
1 Symptoms related to hypermetabolism (e.g. weight loss, lassitude, anorexia or night sweats).
2 Splenomegaly is nearly always present and is frequently massive. In some patients splenic enlargement is associated with considerable discomfort, pain or indigestion.
3 Features of anaemia may include pallor, dyspnoea and tachycardia.
4 Bruising, epistaxis, menorrhagia or haemorrhage from other sites because of abnormal platelet function.
5 Gout or renal impairment caused by hyperuricaemia from excessive purine breakdown may be a problem.
6 Rare symptoms include visual disturbances and priapism.
7 In up to 50% of cases the diagnosis is made incidentally from a routine blood count.
Laboratory findings
1 Leucocytosis is usually >50 × 109/L and sometimes >500 × 109/L (Fig. 14.2). A complete spectrum of myeloid cells is seen in the peripheral blood. The levels of neutrophils and myelocytes exceed those of blast cells and promyelocytes (Fig. 14.3).
2 Increased circulating basophils.
3 Normochromic normocytic anaemia is usual.
4 Platelet count may be increased (most frequently), normal or decreased.
5
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


