Classification of leukaemia
The main classification is into four types: acute and chronic leukaemias, which are further subdivided into lymphoid or myeloid.
Acute leukaemias are usually aggressive diseases in which malignant transformation occurs in the haemopoietic stem cell or early progenitors. Genetic damage is believed to involve several key biochemical steps resulting in (i) an increased rate of proliferation, (ii) reduced apoptosis and (iii) a block in cellular differentiation. Together these events cause accumulation in the bone marrow of early haemopoietic cells known as blast cells. The dominant clinical feature of acute leukaemia is usually bone marrow failure caused by accumulation of blast cells although organ infiltration also occurs. If untreated, acute leukaemias are usually rapidly fatal but, paradoxically, they may be easier to cure than chronic leukaemias.
Acute leukaemia is normally defined as the presence of over 20% of blast cells in the blood or bone marrow at clinical presentation. However, it can be diagnosed with less than 20% blasts if specific leukaemia-associated cytogenetic or molecular genetic abnormalities are present (Table 13.1).
Table 13.1 Classification of acute myeloid leukaemia (AML) according to the WHO classification 2008 (modified).
| Acute myeloid leukaemia with recurrent genetic abnormalities |
| AML with t(8;21)(q22;q22);RUNX1RUNX1T1 |
| AML with inv(16)(p13.1q22) or t(16;6)(p13.1;q22); CBFB-MYH11 |
| AML with t(15;17)(q22;q12); PML-RARA |
| Provisional entity: AML with mutated NPM1 |
| Provisional entity: AML with mutated CEBPA |
| Acute myeloid leukaemia with myelodysplasia related changes |
| Therapy-related myeloid neoplasms (t-AML) |
| Acute myeloid leukaemia, not otherwise specified |
| AML with minimal differentiation |
| AML without differentiation |
| AML with maturation |
| Acute myelomonocytic leukaemia |
| Acute monoblastic/monocytic leukaemia |
| Acute erythroid leukaemia |
| Acute megakaryoblastic leukaemia |
| Acute basophilic leukaemia |
| Acute panmyelosis with myelofibrosis |
| Myeloid sarcoma |
| Myeloid proliferations related to Down syndrome |
| Transient abnormal myelopoiesis |
| Myeloid leukaemia |
The lineage of the blast cells is defined by examination (morphology), immunophenotypic (flow cytometry), cytogenetic and molecular analysis. This will define whether the blasts are of myeloid or lymphoid lineage and also localize stage of cellular differentiation (Table 13.2). The typical ‘myeloid immunophenotype’ is CD13+, CD33+, CD117+and TdT−(Table 13.2; Fig. 13.1) and special antibodies are helpful in the of the rare undifferentiated, erythroid or megakaryoblastic subtypes (Table 13.2).
Table 13.2 Specialized tests for acute myeloid leukaemia
| Cytochemistry | |
| Myeloperoxidase | + (including Auer rods) |
| Sudan black | + (including Auer rods) |
| Non-specific esterase | + in M4, M5 |
| Immunological markers (flow cytometry) | |
| CD13, CD33, CD117 | + |
| Glycophorin | + (erythroid) |
| Platelet antigens (e.g. CD41) | + (megakaryoblastic) |
| Myeloperoxidase | + (undifferentiated) |
Chromosome and genetic analysis (Tables 13.1 & 13.3)
Figure 13.1 Development of three cell lineages from pluripotential stem cells giving rise to the three main immunological subclasses of acute leukaemia. The immunological characterization using pairs of markers is shown, as well as the three markers characterizing the early ‘stem’ cells. AML, acute myeloid leukaemia; B-ALL, B-cell acute lymphoblastic leukaemia; c, cytoplasmic; HLA, human leucocyte antigen; T-ALL, T-cell acute lymphoblastic leukaemia; TdT, terminal deoxynucleotidyl transferase .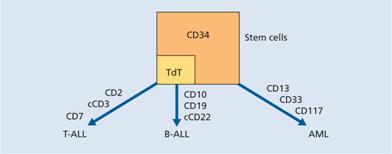
Cytogenetic and molecular analysis is essential and is usually performed on marrow cells although blood may be used if the blast cell count is particularly high. Cytochemistry can be useful in determining the blast cell lineage but is no longer performed in centres where the newer and more definitive tests are available.
Incidence
Acute myeloid leukaemia (AML) is the most common form of acute leukaemia in adults and becomes increasingly common with age with a median onset of 65 years. It forms only a minor fraction (10–15%) of the leukaemias in childhood. Cytogenetic abnormalities and response to initial treatment have a major influence on prognosis (Table 13.3).
Table 13.3 Prognostic factors in acute myeloid leukaemia (AML).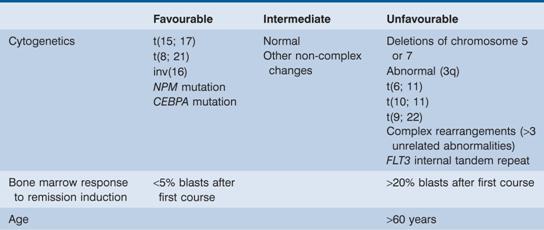
Classification
AML is classified according to the World Health Organization (2008) scheme. There is an increasing focus on the genetic abnormalities within the malignant cells and it is likely that ultimately almost all AML cases will be classified by specific genetic subtype. Currently this is not possible but many genetic subtypes have been determined. Approximately 60% of cases exhibit karyotypic abnormalities on cytogenetic analysis and many cases with a normal karyotype carry mutations in genes such as nucleophosmin (NPM), FLT3 and CEBPA detected only by molecular methods.
Six main groups of AML are recognized (Table 13.1) and these are discussed below.
1 AML with recurrent genetic abnormalities encompasses subtypes with specific chromosomal translocations or gene mutations. The detection of these abnormalities defines the tumour as AML and so the diagnostic criteria for this subgroup are relaxed in that the bone marrow blast cell count does not need to exceed 20% in order to make a diagnosis. In general these disorders have a good prognosis.
2 AML with myelodysplasia-related changes. In this group the AML is associated with microscopic features of dysplasia in at least 50% of cells in at least two lineages. The clinical outcome of these patients is impaired in relation to the first subgroup.
3 Therapy-related myeloid neoplasms (t-AML) arise in patients who have been previously treated with drugs such as etoposide or alkylating agents. They commonly exhibit mutations in the MLL gene and the clinical response is usually poor.
4 AML, not otherwise specified. This group is defined by the absence of cytogenetic abnormalities and comprises around 30% of all cases. Mutations in the NPM and FLT3 genes are seen in approximately 50% and 30% of AML cases, respectively, and are more frequent in those with normal cytogenetics.
5 Myeloid sarcoma is rare but refers to a disease that resembles a solid tumour but is composed of myeloid blast cells.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


