β-Cell Dysfunction in Type 2 Diabetes Mellitus
Jack L. Leahy
Type 2 diabetes mellitus is a worldwide health crisis; the World Health Organization predicts an incidence of 300 million by 2025 (1). The past decade has seen great progress in our understanding of the pathogenesis of type 2 diabetes (Fig. 25.1). The initial event is a genetic predisposition for glucose intolerance. Although specific polymorphisms or mutated genes are not yet known, many that affect the liver, skeletal muscle, adipose, β-cells, or brain physiology will undoubtedly be uncovered. Lifestyle and environmental factors also determine whether glucose intolerance develops (2). An observation from many cross-sectional and longitudinal studies is the presence of insulin resistance (from obesity, a high-fat diet, inactivity, aging, or a genetic basis) early in the course, even predating the hyperglycemia (3,4,5,6,7). This observation is so well known that type 2 diabetes is understood by most students and practicing physicians to be a disease of insulin resistance. However, several lines of investigation, summarized below, have established a crucial role for β-cell dysfunction.
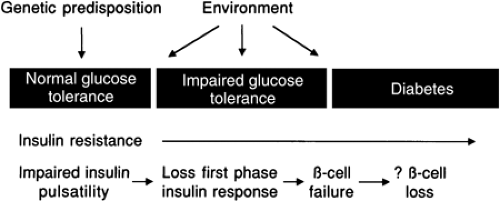 Figure 25.1. Proposed schema for the pathogenesis of type 2 diabetes. Predicted time points for the onset of various aspects of the β-cell dysfunction in this disease that are shown are based on the available literature, as reviewed in the chapter. |
β-Cell dysfunction is always found in type 2 diabetes. Furthermore, it occurs early and likely predates the hyperglycemia (8,9,10).
Insulin resistance changes little during the progression from impaired glucose tolerance (IGT) to diabetes. In contrast, β-cell function undergoes substantial change. Cross-sectional and longitudinal studies have shown an increase in fasting insulin level and in insulin response to oral glucose in the early phases of the disease that keep glycemia normal despite the presence of insulin resistance, followed by a decrease when fasting glycemia surpasses 140 mg/dL (11,12). This inverted U-shaped curve of insulin levels (Fig. 25.2) was initially used to support the importance of insulin resistance in the early stages of the disease, as reflected in the increasing insulin output. However, more recent interest has focused on the decline—the so-called β-cell failure—as it coincides with, and is thought to cause, the progression from IGT to overt diabetes (9,10,13,14,15). Further, prospective study of persons with normal glucose tolerance (16) or IGT (17) showed that those with the lowest insulin response to a meal or to an oral glucose challenge (low insulin responders) carry the highest risk for developing type 2 diabetes later in life. These experimental observations highlight the importance of β-cell function in determining the glycemic status of at-risk persons.
Following the onset of diabetes, the β-cell dysfunction is reversible to a large degree by intensive glycemic control (18,19,20). In one of these studies (20), insulin pumps were used for 21 days in persons with type 2 diabetes. Improvement was observed not only in β-cell function but also in insulin-mediated suppression of hepatic glucose production and in some lessening of insulin resistance. These findings established the concept that part of the diabetes phenotype is acquired from metabolic derangements in the prediabetes/IGT phase of the disease through glucotoxicity (21,22), β-cell exhaustion (23), or less well defined mechanisms. Dysmetabolism-induced acquired tissue dysfunction explains why type 2 diabetes presents a relatively uniform clinical syndrome in different ethnic groups and populations despite presumed diverse genetic causes.
Over time, type 2 diabetes becomes less responsive to oral hypoglycemic therapy in tandem with worsening β-cell function (24,25). The working assumption is that pathogenic elements that lead to a loss in β-cell mass become operative.
There is little information from human studies regarding the biochemical and molecular basis for the β-cell dysfunction in type 2 diabetes because of the unavailability of pancreatic biopsy. Instead, animals—both rodents and larger animals—have been the major venue of investigation, as their β-cell (dys)function with diabetes closely resembles that of humans. Animal studies have identified a panoply of β-cell abnormalities with diabetes. We do not yet know which mechanisms operate in humans, but it seems certain that the β-cell dysfunction will be multifaceted and will entail multiple mechanisms.
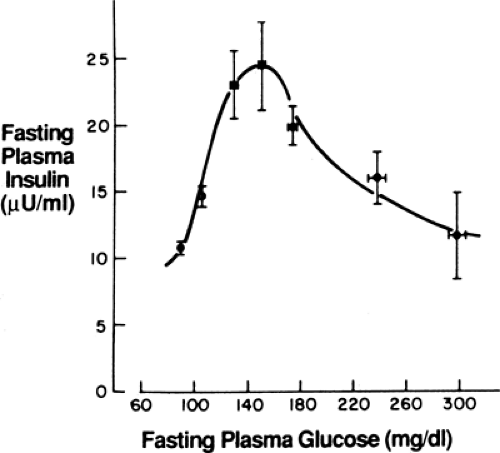 Figure 25.2. Fasting immunoreactive plasma insulin levels in nonobese subjects stratified by the level of glycemia. (From DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in noninsulin-dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism 1989;38:387–395, with permission.) |
To summarize, there is now general acceptance that β-cell dysfunction plays a crucial and necessary role in type 2 diabetes. Indeed, most investigators in the field consider defective insulin secretion and tissue insulin resistance of equal importance in the development of this disease; in the vast majority of affected persons, both must be present for the diabetes syndrome to occur (26). Whether defective insulin secretion and tissue resistance to insulin represent pleiotropic tissue effects of a single defect or multiple abnormalities is unknown. Another understanding is that type 2 diabetes is a progressive disease. Loss of β-cell function, and possibly of β-cell mass, is believed to underlie this progression, highlighting the pivotal role of the β-cell in determining the natural history of this disease. Many excellent reviews on β-cell dysfunction in type 2 diabetes are available (27,28,29,30,31,32).
NORMAL β-CELL FUNCTION
Pancreatic β-cells regulate the storage and metabolism of cellular fuels through their secretion of insulin. This crucial function is accomplished through a feedback loop whereby glycemia upregulates β-cell function—insulin secretion, proinsulin biosynthesis, processing of proinsulin to insulin, and β-cell replication rate—and the secreted insulin in turn lowers glycemia by inhibiting hepatic and renal glucose production and increasing glucose uptake into target organs, primarily skeletal muscle. Glucose regulation of insulin secretion occurs directly (glucose-induced insulin secretion) and also through modulation of the insulin response to insulinotropic hormones, nutrients, and neurotransmitters (glucose potentiation of nonglucose secretagogues; Fig. 25.3). These dual aspects of glucose-regulated insulin secretion are a potent modulatory system that ensures that the tissues’ needs for insulin are exactly met in the fasting and postprandial
states. The need for insulin is for the most part determined by the sensitivity of tissue to insulin—a curvilinear relationship exists with insulin secretion (Fig. 25.4) (33). Thus, even wide variations in insulin sensitivity such as the insulin resistance of puberty (34), pregnancy (35), and aging (36) do not normally affect glycemia. Also, dysfunction of the system can be determined by graphing insulin sensitivity and secretion from the experimental population to see if these values fall on the curvilinear curve (10,37,38). Another in vivo approach to testing the glucose-sensing character of β-cells uses a graded glucose infusion to produce a progressive increase in glycemia (39).
states. The need for insulin is for the most part determined by the sensitivity of tissue to insulin—a curvilinear relationship exists with insulin secretion (Fig. 25.4) (33). Thus, even wide variations in insulin sensitivity such as the insulin resistance of puberty (34), pregnancy (35), and aging (36) do not normally affect glycemia. Also, dysfunction of the system can be determined by graphing insulin sensitivity and secretion from the experimental population to see if these values fall on the curvilinear curve (10,37,38). Another in vivo approach to testing the glucose-sensing character of β-cells uses a graded glucose infusion to produce a progressive increase in glycemia (39).
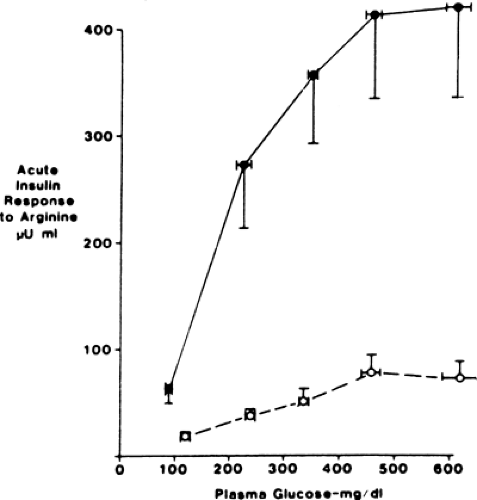 Figure 25.3. Acute insulin responses to 5 g of intravenous arginine at five different glucose levels in eight subjects with type 2 diabetes (open circles) and eight control subjects (closed circles). Insulin responses at all of the glucose levels are markedly attenuated in the subjects with type 2 diabetes. (From Ward WK, Bolgiano DC, McKnight B, et al. Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest 1984;74:1318–1328, with permission from the American Society for Clinical Investigation.) |
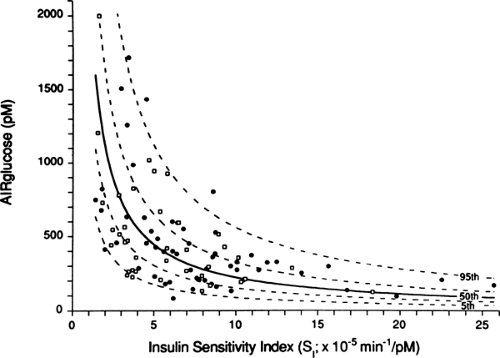 Figure 25.4. Curvilinear relationship between insulin sensitivity (SI) and the first-phase insulin response (AIRglucose) in 93 normoglycemic subjects: 55 men (closed circles) and 38 women (open squares). Lines depicting the 5th, 25th, 50th, 75th, and 95th percentiles are shown. (Copyright © 1993 from the American Diabetes Association. From Kahn SE, Prigeon RL, McCulloch DK, et al. Quantification of the relationship between insulin sensitivity and β-cell function in human subjects: evidence for a hyperbolic function. Diabetes 1993;42:1663–1672, with permission from the American Diabetes Association.) |
It is not only the amount of insulin released that is important. An acute increase in glycemia that can be approximated experimentally with intravenous glucose elicits a large burst of insulin secretion that lasts 5 to 7 minutes (first phase) followed by sustained insulin secretion that lasts for the duration of the hyperglycemia (second phase). Meals also induce a biphasic pattern of insulin secretion, although the phases are less distinct, with the early phase ascribed to the first 30 minutes and the later phase to the remaining postprandial hyperinsulinemia (1 to 2 hours normally). The biphasic pattern is necessary for normal mealtime glucose tolerance, with the concept being that the first phase primes the insulin-sensitive tissues for the coming food. Supporting this notion are studies that experimentally disrupted the early insulin response to a meal in otherwise healthy, normoglycemic subjects, causing impaired insulin-mediated tissue glucose disposal and excessive postprandial glycemia (40,41).
Insulin secretion occurs as oscillatory pulsations with a periodicity of 11 to 14 minutes, thought to be necessary to fully regulate hepatic glucose production (42,43). Also, large bursts of insulin release (ultradian oscillations) occur several times a day, particularly with meals (44), increasing the efficiency of nutrient clearance following meals (45).
Thus, the β-cell functions in a highly complex fashion that regulates the timing and overall insulin response to a meal to preserve normoglycemia. Quantitative insulin release and the pulsatile patterns can be tested in vivo by means of frequent insulin measurements to an appropriate stimulus. Also, the glucose-sensing and pulsatile-secretion characteristics can be jointly tested with an oscillatory glucose infusion that causes small increases and decreases in plasma glucose levels. Insulin secretion normally attains an oscillatory pattern termed entrainment (46,47). Failure of entrainment has been identified as an early defect in insulin secretion that precedes abnormal responses to more traditional tests. In vivo testing has been aided by several technical advances over the last few years. Insulin-specific assays are now widely available that have eliminated the cross-reactivity with proinsulin and its conversion intermediates (which are biologically inactive) that affected earlier assays. Also, insulin is secreted into the portal vein and undergoes a substantial first-pass clearance by the liver (approximately 50%). Thus, insulin levels in the peripheral circulation only approximate insulin secretion. Many investigators now analyze C-peptide values—the portion of proinsulin that is removed as it is converted to insulin. C-peptide is secreted with insulin in an equimolar ratio but undergoes minimal hepatic degradation and thus can be used for calculating true rates of insulin secretion (48,49).
β-CELL DYSFUNCTION IN TYPE 2 DIABETES
β-Cell Unresponsiveness to Glucose
The distinctive β-cell defect in type 2 diabetes is the loss of the first phase of glucose-induced insulin secretion (Fig. 25.5). This observation was first made in the late 1960s, when persons with
type 2 diabetes were noted to have a delayed insulin response to intravenous glucose, and later was recognized as loss of the first phase. The second phase is also impaired but to a lesser degree (50,51). Subsequent investigation showed that this defect was fully established by the time the fasting glucose level reached 115 mg/dL (52), clearly predating overt diabetes, but was not present in persons with truly normal glucose levels in whom type 2 diabetes developed later (3,6). Thus, it first appears in the prediabetes state, termed IGT, which is clinically manifest as excess postprandial excursions of glycemia. Given the importance of the first-phase insulin response for normal prandial glucose tolerance (40,41), it follows that this defect is an important cause of the IGT state. Attesting to this idea is a study that simulated a burst of insulin with a short-term insulin infusion at the beginning of a meal in persons with type 2 diabetes, which resulted in a marked improvement in postprandial glycemia (53).
type 2 diabetes were noted to have a delayed insulin response to intravenous glucose, and later was recognized as loss of the first phase. The second phase is also impaired but to a lesser degree (50,51). Subsequent investigation showed that this defect was fully established by the time the fasting glucose level reached 115 mg/dL (52), clearly predating overt diabetes, but was not present in persons with truly normal glucose levels in whom type 2 diabetes developed later (3,6). Thus, it first appears in the prediabetes state, termed IGT, which is clinically manifest as excess postprandial excursions of glycemia. Given the importance of the first-phase insulin response for normal prandial glucose tolerance (40,41), it follows that this defect is an important cause of the IGT state. Attesting to this idea is a study that simulated a burst of insulin with a short-term insulin infusion at the beginning of a meal in persons with type 2 diabetes, which resulted in a marked improvement in postprandial glycemia (53).
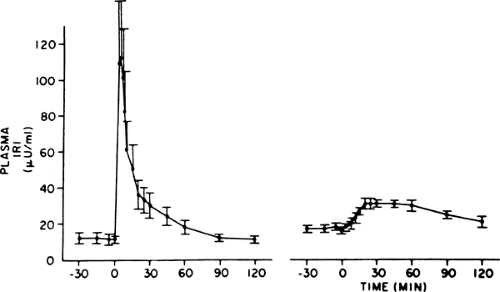 Figure 25.5. Plasma immunoreactive insulin response to 20-g bolus of intravenous glucose in nine subjects with type 2 diabetes (right) and nine normoglycemic control subjects (left). The first-phase insulin response (from 0 to 10 minutes) is totally lacking in the subjects with type 2 diabetes. In contrast, the second phase (from 10 minutes continuing for the duration of hyperglycemia) is intact in the subjects with diabetes and is greater than in the controls because of the persistent hyperglycemia in the subjects with diabetes following the glucose administration. (Reprinted from Pfeifer MA, Halter JB, Porte D Jr. Insulin secretion in diabetes mellitus. Am J Med 1981;70:579–588, with permission from Elsevier Science.) |
A major advance in our understanding occurred with the demonstration that intensive glycemic control restored the first-phase insulin response in subjects with type 2 diabetes (19). At about the same time, glucose-induced insulin secretion was shown to be impaired in diabetic animals (discussed subsequently). An important observation, made in diabetic rats, is that phloridzin reverses the defect (54). Phloridzin promotes glycosuria; it is used experimentally in diabetic animals to restore normoglycemia without changing insulinemia, thus helping to identify pathogenic effects of hyperglycemia. The understanding that has evolved from these findings is that the defect in glucose-induced insulin secretion occurs when β-cells are exposed to a “toxic” environment of an abnormally high level of glycemia (so-called glucose toxicity).
Insulin responses to nonglucose secretagogues are less impaired than those to glucose. When the defective glucose-induced insulin secretion was first investigated, nonglucose agents were thought to be unaffected, a finding that led to the idea that the glucose response was uniquely deranged (selective glucose unresponsiveness). However, later studies that were more carefully controlled for glycemia in the subjects with and without diabetes made it clear that responses to nonglucose secretogogues also were impaired, although less so than those to glucose. This challenged the concept that glucose unresponsiveness is the prototypical β-cell abnormality in type 2 diabetes; however, subsequent investigation (55) showed that the basis for the defective nonglucose-mediated secretion was impaired glucose potentiation (Fig. 25.3). The time of onset for the glucose potentiation defect is not as well defined as that for glucose-induced insulin secretion, and it is not clear if these dual abnormal effects of glucose on insulin secretion represent a single defect or separate biochemical/molecular defects in glucose sensing by the β-cell. Viewed together, type 2 diabetes entails defective glucose regulation of insulin secretion through both pathways—glucose-induced insulin secretion (in particular the first phase) and glucose potentiation—emphasizing why fasting and postprandial hyperglycemia are defining characteristics of this disease.
For many years there was confusion about how β-cell dysfunction could be present in the prediabetes/IGT state, in particular the loss of the first-phase response, when countless studies had showed hyperinsulinemia (both fasting and after a glucose challenge or a meal) at that time (Fig. 25.2). Insight into this seeming paradox came with the understanding of the importance of the early insulin response for control of postprandial glycemia (40,41); loss of this early response results in an excessive meal-induced rise in glycemia, and this hyperglycemia causes the late insulin response to exceed that seen normally. Previous studies had generally looked at insulin levels 2 hours after a meal or oral glucose challenge and thus had missed the defect in early insulin secretion. This concept is shown in Figure. 25.6, which shows the 30-minute and 2-hour insulin values after oral glucose challenge across a wide range of glycemia (29). Note the disparity as the early insulin response decreases with increasing glycemia at a time when later insulin release is increasing.
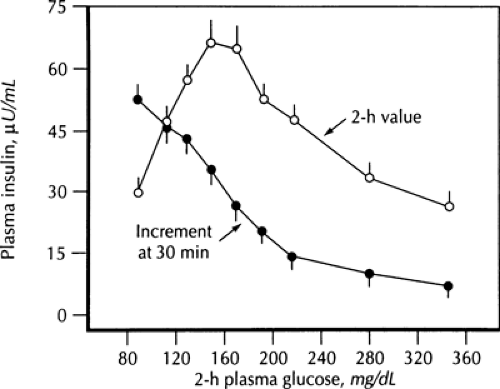 Figure 25.6. Comparison of 30-minute and 2-hour plasma insulin responses during oral glucose tolerance tests as a function of 2-hour plasma glucose values in 294 subjects. (From Gerich J, Van Haeften T. Insulin resistance versus impaired insulin secretion as the genetic basis for type 2 diabetes. Curr Opin Endocrinol Diabetes 1998;5:144–148, with permission.) |
Thus, a characteristic feature of type 2 diabetes is loss of the first-phase insulin response to a meal. It occurs early in the
course of type 2 diabetes, predating fasting hyperglycemia, and is a major cause of the postprandial hyperglycemia that characterizes IGT and overt diabetes. This concept underlies the recent use of drugs for type 2 diabetes (meglitinides and phenylalanine derivatives) that are taken at meals to induce a rapid, short-lived insulin response.
course of type 2 diabetes, predating fasting hyperglycemia, and is a major cause of the postprandial hyperglycemia that characterizes IGT and overt diabetes. This concept underlies the recent use of drugs for type 2 diabetes (meglitinides and phenylalanine derivatives) that are taken at meals to induce a rapid, short-lived insulin response.
Abnormal Pulsatile Secretion of Insulin
The pattern of orderly oscillations of insulin secretion with an 11- to 14-minute periodicity is lost in type 2 diabetes (56). Relatives of persons with type 2 diabetes show this defect when their glucose tolerance is normal (57,58), showing that it occurs early in the course of the disease. This might imply a genetic etiology. Countering that idea is a recent study showing recovery of oscillations in persons with type 2 diabetes after an overnight infusion of somatostatin, which pharmacologically blocks insulin release—interpreted by the authors as indicating that depletion of a readily releasable pool of insulin granules underlies the oscillation defect (59).
It is now generally accepted that the abnormal insulin pulsatility impairs normal regulatory control of insulin over hepatic glucose production. However, this understanding is relatively recent. The importance of the pulsatility defect was uncertain when first observed, as the magnitude of the pulsations in the normoglycemic control subjects was quite small (56): It was difficult to appreciate that such a minor effect would have any physiologic importance. Those studies used blood from the peripheral circulation. Insulin is secreted into the portal vein and undergoes substantial hepatic extraction. The breakthrough came with the understanding that the pulses are in fact large and are damped by hepatic extraction so that only a small fraction escapes the liver (42,60). Thus, the abnormal peripheral pulsations in type 2 diabetes represent very distorted insulin delivery to the liver, fitting with the idea of major dysregulation to the insulin-hepatic system. Future treatments might target this abnormality. For example, glucagon-like peptide-1 (GLP-1), which is under investigation as a hypoglycemic therapy for type 2 diabetes, increases pulsatile insulin secretion (61).
The ultradian oscillations—the large bursts of insulin that occur every 1 to 2 hours and more frequently with meals—also are disrupted in type 2 diabetes (62,63). This fact is well established, but the impact on glucose homeostasis is not totally clear. Regardless, pulsatile delivery of insulin holds a physiologic advantage, as evidenced by studies that have shown a greater hypoglycemic effect of pulsed versus continuously infused exogenous insulin (64). As such, loss of the pulsatile pattern of insulin secretion in type 2 diabetes disrupts this aspect of the glucose homeostasis system.
Increased Proinsulin-to-Insulin Ratio
The blood levels of insulin precursors (proinsulin and its conversion intermediates, which have only weak biologic activity) are disproportionately increased relative to insulin in type 2 diabetes (65,66). The same observation has been made in patients with diabetes caused by cystic fibrosis (67), a finding that raises the possibility that this disproportionate increase is another manifestation of hyperglycemia-induced β-cell dysfunction. However, the relationship of this increased ratio to hyperglycemia has been confused by seemingly inconsistent data. Most cross-sectional data show that the increased proinsulin/insulin ratio in type 2 diabetes occurs after the onset of hyperglycemia (68,69), with the ratio increasing as glycemia worsens (69,70). In disagreement are reports of an increased proinsulin/insulin ratio in the absence of glucose intolerance in populations at high risk for type 2 diabetes (71,72). Treatment studies to determine how reversal of hyperglycemia affects the proinsulin/insulin ratio also have not clarified its dependence on abnormal glycemia, because both improvement (73) and no effect (74) have been reported. Insight into this conundrum has come from the previously mentioned study that administered an overnight infusion of somatostatin to persons with type 2 diabetes; the proinsulin/insulin ratio was normalized by this pharmacologic inhibition of insulin secretion (59). This observation suggests that excessive insulin secretion, as opposed to hyperglycemia per se, underlies the raised proinsulin/insulin ratio. Another study of type 2 diabetes (75) and a study in diabetic sand rats (76) made a similar conclusion.
Our laboratory has studied diabetic rats in an effort to gain insight into the mechanistic basis for the raised proinsulin/insulin ratio (77,78,79). We first studied rats that had 90% of their pancreas removed (90% pancreatectomy) and observed an increased abundance of proinsulin-like peptides relative to insulin in pancreatic extracts, suggesting that the underlying defect was the storage, and subsequent secretion, of material enriched in proinsulin (77). We made the same observation in normal rats that received 48-hour infusions of glucose (78). That study showed the increased ratio resulted from a decreased insulin content, not from an increased proinsulin content (Fig. 25.7). Also, co-infusion of diazoxide to inhibit insulin secretion blocked the increase in the proinsulin/insulin ratio, results analogous to those seen in humans infused with somatostatin (59). These findings are in accord with the previously stated concept that an ongoing hypersecretion of insulin leading to depletion of the releasable insulin stores is the cause of the enhancement in proinsulin secretion, likely through secretion of immature, proinsulin-enriched granules. Confirmatory evidence was obtained by infusing normal rats for 3 days with large amounts of the insulin secretagogue tolbutamide (plus glucose to keep them euglycemic) and observing a raised percentage of proinsulin in pancreatic extracts (78). Furthermore, this concept implies that there is no biochemical or molecular defect in the proinsulin-processing pathway in diabetes, which we demonstrated to be the case in glucose-infused rats (79).
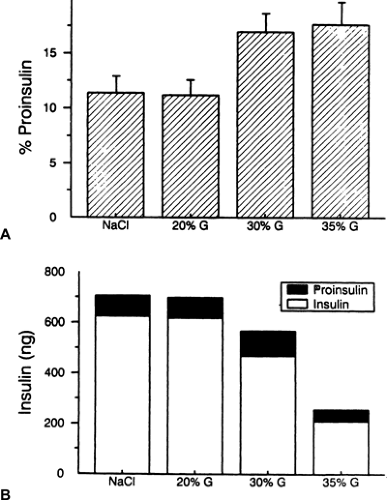 Figure 25.7. Relative proportion of proinsulin in pancreatic extracts from rats that received a 48-hour infusion with glucose. Pancreases underwent extraction, insulin/proinsulin precipitation, and separation of the insulins and proinsulins by high-performance liquid chromatography. Immunoreactive insulin and proinsulin were determined as the areas under the curve of the different peaks measured by insulin radioimmunoassay. A: % proinsulin = plasma immunoreactive insulin response (IRI) proinsulin/(IRI insulin + IRI proinsulin). B: IRI insulin and proinsulin in chromatography samples. Note that an increase in % proinsulin resulted from a decrease in insulin content not from an increase in proinsulin content. (Copyright © 1993, American Diabetes Association. From Leahy JL. Increased proinsulin/insulin ratio in pancreas extracts of hyperglycemic rats. Diabetes 1993;42:22–27. Reprinted with permission from the American Diabetes Association.) |
Thus, the increased proportion of stored and circulating proinsulin in the diabetic rats appears to be secondary to a hypermobilization of granules, which leads to a rapid transit time and thus to incomplete processing to fully mature insulin. This hypersecretion scenario fits perfectly with the findings in patients with type 2 diabetes (59,75). However, enhanced insulin secretion per se is not enough to raise the proinsulin/insulin ratio, as is apparent from studies of nondiabetic obese subjects who are hyperinsulinemic but who have normal to lowered proinsulin/insulin ratios (68,80). Instead, some underlying element of β-cell dysfunction must be present as well.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


