Thyroid Hormone Synthesis
Peter Kopp
Thyroid hormone biosynthesis, storage, and secretion require a series of highly regulated steps. Iodide, the rate-limiting substrate for thyroid hormone synthesis, is actively transported into thyrocytes by the sodium-iodide symporter (NIS/SLC5A5) at the basolateral membrane (Fig. 4B.1). This transport is dependent on the electrochemical gradient generated by the Na+, K+-ATPase. At the apical membrane of the cells, iodide efflux into the follicular lumen appears to be mediated, in part, by pendrin (PDS/SLC26A4) in conjunction with at least one other channel. On the luminal side of the apical membrane, iodide is oxidized by thyroid peroxidase (TPO), a reaction that requires the presence of hydrogen peroxide (H2O2). H2O2 is generated by the calcium-dependent flavoprotein NADPH oxidase DUOX2, an enzyme that requires a specific maturation factor DUOXA2. In the follicular lumen, thyroglobulin serves as matrix for the synthesis of thyroxine (T4) and triiodothyronine (T3). In a first step, TPO iodinates selected tyrosyl residues on thyroglobulin, a process referred to as organification or iodination. This results in the formation of mono- and diiodotyrosines (MIT, DIT). In the subsequent coupling reaction, which is also catalyzed by TPO, two iodotyrosines are coupled to form T4 or T3. Iodinated thyroglobulin is stored as colloid in the follicular lumen. In response to demand for thyroid hormone secretion, thyroglobulin is internalized into the follicular cell by micro- and macropinocytosis, and digested in lysosomes. Subsequently, the thyronines T4 (∼80%) and T3 (∼20%) are released into the blood stream. The secretion of thyroid hormones at the basolateral membrane occurs, in part, through the monocarboxylate channel MCT8 (SLC16A2). MIT and DIT are deiodinated by the iodotyrosine dehalogenase DEHAL1, and the released iodide is recycled for hormone synthesis.
Thyroid hormone synthesis is dependent on the nutritional availability of iodide, and it is predominantly regulated by thyrotropin (TSH). TSH binds to its cognate receptor, a member of the G-protein–coupled seven transmembrane receptors, which is expressed at the basolateral membrane (Fig. 4B.1) (see section on the TSH receptor in Chapter 10B). Binding of TSH to its receptor leads primarily to coupling to Gsα and subsequent activation of adenylyl cyclase. The resulting increase in cyclic AMP formation leads to phosphorylation of protein kinase A and to activation of targets in the cytosol and the nucleus. The TSH-dependent cyclic AMP cascade is the major regulator of growth, differentiation, and hormone secretion of thyrocytes. At higher concentrations of TSH, stimulation of Gq/11 and the phospholipase C-dependent inositol phosphate Ca2+/diacylglycerol pathway activates H2O2 generation and iodination.
In addition to TSH, iodide uptake is inversely regulated by the intracellular iodide concentration, and the organification process is transiently blocked by high intracellular iodide concentrations (Wolff–Chaikoff effect). These autoregulatory mechanisms protect the thyroid from iodide excess while ensuring adequate iodide uptake for hormone synthesis.
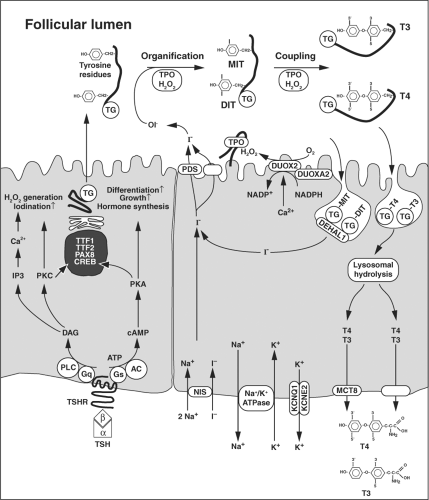 Figure 4B.1 Thyroid hormone synthesis secretion and major signaling pathways in thyrocytes. AC, adenylyl cyclase; cAMP, cyclic AMP; DAG, diacylglycerol; DEHAL1, iodotyrosine dehalogenase 1; DIT, diiodotyrosine; DUOX, dual oxidase 2; DUOXA2, dual oxidase maturation factor 2; KCNQ1 and KCNE2, potassium channel subunits; MCT8, monocarboxylate transporter 8; MIT, monoiodotyrosine; NIS, sodium/iodide symporter; PDS, pendrin (SLC26A4, solute carrier 26A4); PKA, protein kinase A; PLC, phospholipase C; TG, thyroglobulin; TPO, thyroid peroxidase; TSH, thyrotropin; TSHR, TSH receptor. |
Iodide Uptake into the Thyroid
Under physiological conditions, the thyroid iodide concentration is 20 to 40 times higher than the plasma iodide concentration, and iodide uptake occurs against a cell to plasma electrochemical gradient of about -40 mV (1). The dependence of iodide uptake on a sodium gradient created by Na+, K+-ATPase led to the prediction that iodide uptake is mediated by a Na+/I– symporter (1,2,3,4,5), and was confirmed by the cloning of the sodium-iodide symporter NIS, which mediates iodide uptake at the basolateral membrane (4,5,6). The properties of NIS are discussed in more detail in Chapter 4A.
The Gene and Protein Structure of the Sodium/Iodide Symporter
The ability of thyroid cells to concentrate iodide was recognized as early as 1896 (7), a century before the cloning of NIS (6,8). Human NIS is encoded by a single copy gene with 15 exons that is located on chromosome 19p13 (9). NIS, officially designated as Solute Carrier 5A (SLC5A), belongs to a family of transporters that relies on an electrochemical sodium gradient as the driving force for solute transport. Human NIS is a 643–amino acid protein. Current information about its secondary structure predicts 13 transmembrane domains with an extracellular amino terminus and an intracellular carboxy terminus (10,11). Mature human NIS is a glycoprotein of ∼108 kDa (11). Partial or total deglycosylation does not impair activity, stability, or membrane targeting of NIS (11). NIS appears to exist primarily as a dimer stabilized by a disulfide bridge (12).
 Figure 4B.2 Schematic structure of the NIS (sodium/iodide symporter), TG (thyroglobulin), and TPO (thyroid peroxidase) gene promoters. (From Dohán O, De la Vieja A, Paroder V, et al. The sodium/iodide symporter (NIS): characterization, regulation, and medical significance. Endocr Rev 2003;24:48, and Damante G, Di Lauro R. Thyroid-specific gene expression. Biochim Biophys Acta 1994;1218:255, modified with permission.) |
Functional Characterization of the Sodium/Iodide Symporter
Consistent with findings obtained in FRTL-5 cells (3), the Michaelis–Menten constant (Km) of NIS for iodide expressed in Xenopus oocytes is ∼36 μM (6). Electrophysiologic studies in oocytes demonstrate that the transport of iodide by NIS is electrogenic, resulting in an inwardly directed influx of positive
charge upon addition of iodide to the extracellular perfusing solution (13). The inward steady-state current is due to the influx of sodium, and the stoichiometry of sodium to iodide is 2:1 (13). On the basis of kinetic results, it is thought that sodium binds to NIS before iodide, and that this is followed by a simultaneous transport of both ions (13).
charge upon addition of iodide to the extracellular perfusing solution (13). The inward steady-state current is due to the influx of sodium, and the stoichiometry of sodium to iodide is 2:1 (13). On the basis of kinetic results, it is thought that sodium binds to NIS before iodide, and that this is followed by a simultaneous transport of both ions (13).
Both perchlorate and thiocyanate cause rapid release of intracellular iodide across the basolateral membrane if organification is blocked by propylthiouracil (PTU) (14,15). This phenomenon is the basis for the perchlorate discharge test, which is used to evaluate intrathyroidal iodide organification (15). Perchlorate and thiocyanate inhibit iodide uptake through competitive inhibition of NIS (13). In contrast to thiocyanate, perchlorate does not elicit an inward current, a finding that led to a debate whether perchlorate is transported by NIS despite its potent inhibitory action on iodide uptake (13,16,17). However, several studies have now provided evidence that perchlorate is also transported by NIS. Studies using tetrahedral oxyanions in transfected cells inferred that NIS might mediate uptake of perchlorate (18). Subsequent studies in transfected cells and mammary cells in vivo provided evidence that perchlorate is actively transported by NIS (19). Kinetic analyses with a structurally related anion, perrhenate (ReO4–) revealed that its transport is electroneutral, suggesting that the NIS-mediated sodium/perchlorate transport stoichiometry is also electroneutral (19). Direct evidence for NIS-mediated perchlorate transport was also obtained by measuring the anion directly with a sensitive ion chromatography tandem mass spectrometry method (20).
NIS function is dependent on the electrochemical gradient generated by the Na+, K+-ATPase. At the basolateral membrane, this also requires the presence of a constitutively active potassium channel consisting of the KCNQ1 and KCNE2 subunits promoting K+ efflux (21,22). Targeted disruption of the Kcne2 subunit results in hypothyroidism with secondary dwarfism, alopecia, goiter and cardiac abnormalities (21). Similarly, Kcnq1 knockout mice also have a hypothyroid phenotype (22).
Regulation of Iodide Uptake and Sodium/Iodide Symporter Function and Expression
TSH, acting through the cyclic AMP pathway, stimulates iodide accumulation in the thyroid (23,24). The increase in iodide uptake is the consequence of increased transcription of NIS and posttranscriptional stimulation of NIS activity. TSH upregulates NIS mRNA and protein expression in vivo and in vitro (10,25,26,27). Consistent with an expression pattern that is not restricted to the thyroid, the structure of the promoter region of the NIS gene differs from those of the TG (thyroglobulin) and TPO (thyroperoxidase) genes (Fig. 4B.2) (28,29,30).
TSH also modulates NIS protein turnover; in the presence of TSH, the half-life of NIS in FRTL-5 cells is ∼5 days and in its absence it decreases to ∼3 days (31). Aside from increasing NIS synthesis, TSH also regulates posttranslational events such as the subcellular distribution of NIS, specifically the targeting to and retention of NIS in the basolateral membrane. In the absence of TSH, NIS remains in the cell, and little reaches the plasma membrane.
In addition to the regulation by TSH, iodide accumulation and organification are directly regulated by iodide itself (32,33,34). Negative regulation of the expression of NIS mRNA occurs in thyroid tissue of dogs fed moderate doses of iodide administered for 2 days (35). High doses of iodide block thyroid hormone synthesis through inhibition of the organification process, the Wolff–Chaikoff effect (32). This transient blockade is dependent on the intracellular iodide concentration (32,33).
The escape from the acute Wolff–Chaikoff effect involves a decrease in iodide transport, which leads to intracellular iodide concentrations that are too low to maintain the inhibitory effect (36). At the molecular level, an iodide-induced downregulation of NIS expression, possibly an increase in NIS protein turnover, and a decrease in NIS activity contribute to this complex autoregulatory phenomenon induced by an increase in intracellular iodide concentrations (35,37,38,39). The downregulation of NIS expression and activity is thyroid-specific, independent of TSH, and may be associated with formation of inactive NIS dimers. Thyroglobulin has been reported to downregulate NIS transcription (40,41), as do several cytokines including transforming growth factor-β, tumor necrosis factor-α, interleukin-1, and interferon-γ (5,42).
The escape from the acute Wolff–Chaikoff effect involves a decrease in iodide transport, which leads to intracellular iodide concentrations that are too low to maintain the inhibitory effect (36). At the molecular level, an iodide-induced downregulation of NIS expression, possibly an increase in NIS protein turnover, and a decrease in NIS activity contribute to this complex autoregulatory phenomenon induced by an increase in intracellular iodide concentrations (35,37,38,39). The downregulation of NIS expression and activity is thyroid-specific, independent of TSH, and may be associated with formation of inactive NIS dimers. Thyroglobulin has been reported to downregulate NIS transcription (40,41), as do several cytokines including transforming growth factor-β, tumor necrosis factor-α, interleukin-1, and interferon-γ (5,42).
Mutations in the Sodium/Iodide Symporter Gene
Congenital hypothyroidism can be caused by developmental defects of the thyroid, thyroid dysgenesis, or inborn errors of metabolism in one of the steps required for thyroid hormone synthesis, dyshormonogenesis (Chapters 36 and 54) (44,45). Among the patients with dyshormonogenesis, a few have an iodide trapping defect (OMIM# 274400) (46). These patients have little or no uptake of radioiodide in scintigraphic studies both in the thyroid and the salivary gland, and a decreased saliva/plasma radioiodide ratio (46,47). The thyroid may be normal in size at birth, but it enlarges soon thereafter unless the infant is given T4 therapy. After the NIS gene was cloned, several patients with hypothyroidism caused by this defect were found to be homozygous or compound heterozygous for inactivating NIS mutations (4,5,48,49). There is marked heterogeneity among patients with the same NIS mutation, as well as in patients with different mutations in terms of severity and onset of hypothyroidism and goiter development, indicating that modifiers such as nutritional iodide intake have an impact on the phenotype (50). Some of the mutations decrease NIS function by substituting key functional amino acid residues (51), others lead to misfolding and retention of NIS in intracellular compartments (see the preceding section of this chapter) (49,52).
Iodide Efflux from the Thyroid
Compared with the mechanisms mediating iodide uptake at the basolateral membrane, iodide efflux at the apical membrane is less well characterized (Fig. 4B.1). Iodide efflux is stimulated by TSH in both poorly polarized FRTL-5 cells (53), and polarized porcine thyrocytes follicular cells (54,55). In primary porcine thyrocytes grown in a transwell system, bidirectional measurements indicate that TSH stimulates iodide efflux at the apical membrane, while efflux in the basal direction does not change (55). Hence, this rapid effect of TSH facilitates the vectorial transport of iodide into the follicular lumen.
Electrophysiologic studies performed with inverted plasma membrane vesicles suggest the existence of two apical iodide channels that could be involved in iodide efflux (56). One of these channels has a high permeability and specificity for iodide (Km ∼70 μM), while the second channel has a lower affinity for iodide (Km ∼33 mM) (56). However, the identity of these channels has not been established at the molecular level. The demonstration of iodide transport by the anion channel pendrin, together with the phenotype in patients with Pendred’s syndrome, suggest that pendrin is one of the channels promoting apical iodide efflux (57,58,59,60,61). Apical iodide efflux is, however, also possible in the absence of pendrin indicating the existence of at least another iodide transporting entity (62,63,64).
Pendred’s Syndrome and Pendrin (PDS/SLC26A4)
Pendred’s syndrome is an autosomal recessive disorder defined by sensorineural deafness, goiter, and impaired iodide organification (OMIM# 274600) (65,66). After linking Pendred’s syndrome to chromosome 7q22–31.1 (67,68), the PDS gene, now officially designated as SLC26A4 (Solute Carrier 26A4), was cloned (57). The PDS/SLC26A4 gene encompasses 21 exons and contains an open reading frame of 2,343 bp (Fig. 4B.3). The SLC26A family contains several multifunctional anion transporters, and the motor protein prestin (SLC26A5), which is expressed in cochlear outer hair cells (69,70,71). The genes encoding pendrin, prestin (SCLC26A5), and DRA/CLD (downregulated in adenoma/congenital chloride diarrhea, SLC26A3) are located in close vicinity on chromosome 7q21–31 and have a very similar genomic structure, suggesting a common ancestral gene.
 Figure 4B.3 Chromosomal location and structure of the PDS/SLC26A4 gene, and current model of the secondary structure of the pendrin (PDS) protein. STAS domain, sulfate transporter and anti-sigma factor antagonist domain; PKA site, protein kinase A site. |
Protein Structure of Pendrin
Pendrin is a highly hydrophobic membrane protein consisting of 780 amino acids (Fig. 4B.3) (57). Initial predictions of the putative transmembrane segments of pendrin proposed a model with 11 transmembrane domains with an intracellular amino terminus and an extracellular carboxy terminus (57). Subsequent studies have shown that both the amino- and the carboxy terminus are located intracellularly and have suggested a model with 12 transmembrane domains (61,72). An alternative model with an extracellular amino terminus and 15 transmembrane domains has also been proposed (73). Human pendrin is a glycoprotein with three putative extracellular N-glycosylation sites (72,74). In extracts of human thyroid membranes studied under denaturing conditions, pendrin is a single molecular species of ∼110 to 115 kDa; deglycosylation leads to a reduction in molecular mass to ∼85 kDa (74). In murine pendrin, glycosylation does not seem to be of importance for membrane insertion and basal activity in terms of chloride/bicarbonate exchange, but elimination of the glycosylation sites disrupts the sensitivity of mouse pendrin to extracellular pH, which appears to increase pendrin activity at low pH through proton regulatory sites (75).
Like other members of the SLC26A family, pendrin contains an STAS (sulfate transporter and anti-sigma factor antagonist) domain in its carboxy terminus (Fig. 4B.3) (76,77). This domain shares similarity with the bacterial anti-sigma factor
antagonists, which are involved in the regulation of sigma factors (σ) that bind to RNA polymerase to confer target gene specificity (76,77). The exact role of the STAS domain in SLC26A family members has not been elucidated; it may play a role in nucleotide binding and/or interactions with other proteins (76,78,79,80,81). The intracellular carboxy terminus contains a protein kinase A consensus site. The insertion of pendrin into the plasma membrane is stimulated by TSH and results in increased iodide efflux, a process that appears to be dependent on phosphorylation of the protein kinase site (82).
antagonists, which are involved in the regulation of sigma factors (σ) that bind to RNA polymerase to confer target gene specificity (76,77). The exact role of the STAS domain in SLC26A family members has not been elucidated; it may play a role in nucleotide binding and/or interactions with other proteins (76,78,79,80,81). The intracellular carboxy terminus contains a protein kinase A consensus site. The insertion of pendrin into the plasma membrane is stimulated by TSH and results in increased iodide efflux, a process that appears to be dependent on phosphorylation of the protein kinase site (82).
Expression and Regulation of Pendrin
Pendrin mRNA, assessed by Northern analysis, is primarily detected in the thyroid and the kidney (57); by RT-PCR it can be detected in many other tissues such as the inner ear, the airways, the lactating mammary gland, the placenta, the endometrium, the testis, the liver and airway epithelial cells (83,84,85,86,87,88,89,90,91,92).
Immunolocalization studies reveal that pendrin is located at the apical membrane of thyrocytes (72,93). The immuno-staining is heterogeneous within and among normal thyroid follicles (93). In thyroid tissue from patients with Graves’ disease, immunostaining revealed higher pendrin protein expression than found in normal thyroid tissue, suggesting a correlation between pendrin abundance and increased iodide organification (72,94). In hyperfunctioning adenomas, the levels of pendrin mRNA were similar, but immunostaining and Western blot analysis revealed more pendrin, as compared with normal thyroid tissue (93,95). In follicular adenomas, the levels of pendrin mRNA and protein were similar to those in normal thyroid tissue (74,96). In hypofunctioning adenomas, the levels of pendrin mRNA were normal, but the pendrin content of the adenomas, as detected by immunostaining, was highly variable (93,95).
The effects of TSH on pendrin mRNA expression in thyroid cell lines has provided results that are, in part, controversial. In FRTL-5 cells, low concentrations of thyroglobulin, but not TSH, insulin, interferon-γ, or iodide, increased pendrin mRNA levels (72). This contrasts with the negative regulation of mRNA expression levels of the NIS, TPO, TG, and TSH– receptor genes after exposure to thyroglobulin in this
cell line (40,41). However, others have reported an increase in SLC26A4 mRNA by RT-PCR after treatment of FRTL-5 cells with TSH and forskolin (97). In PCCL3 cells, pendrin mRNA and protein levels were found to increase after exposure to high doses of TSH, forskolin, and 8-bromo-cyclic AMP (98).
cell line (40,41). However, others have reported an increase in SLC26A4 mRNA by RT-PCR after treatment of FRTL-5 cells with TSH and forskolin (97). In PCCL3 cells, pendrin mRNA and protein levels were found to increase after exposure to high doses of TSH, forskolin, and 8-bromo-cyclic AMP (98).
In differentiated thyroid carcinomas, pendrin mRNA and protein expression were low (74,93,94,99), and did not correlate with NIS expression (99,100). Targeting of pendrin to the plasma membrane was found to be decreased in well-differentiated thyroid cancers (101). Pendrin mRNA was also scarce in thyroid cancer cell lines (72). Consistent with this low or absent expression, aberrant hypermethylation of the promoter region of the SLC26A4 gene was found in the majority of thyroid cancers (102).
Limited analysis of the 4.2 kb 5′-flanking region in renal HEK293, thyroid LA2, and inner ear VOT36 epithelial cell lines suggest increased promoter activity under alkaline conditions and a decrease with an acidic pH (103). Moreover, acidic pH and aldosterone decreased, and alkaline pH increased, endogenous pendrin mRNA level in HEK293 kidney cells (103). The regulation of the pendrin promoter in thyroid cells has not been characterized in further detail. Mice with targeted disruption of the winged helix/forkhead Foxi1 transcription factor have an inner ear phenotype that is similar to the findings in humans and mice with biallelic inactivation of the pendrin gene (104). Foxi1 has been shown to be an upstream regulator of the pendrin promoter (104). A role of FOXE1 is further corroborated by the finding of a patient with deafness associated with an enlarged vestibular aqueduct and digenic mutations in the SLC26A4 and the FOXE1 gene (105).
 Figure 4B.4 Polarized MDCK cells grown in a bicameral system and expressing NIS, PDS (SLC26A4), or NIS and PDS were exposed to a solution containing 125iodide in the lower chamber. There was a large increase in intracellular iodide in the cells expressing NIS. In cells expressing NIS and PDS, the intracellular iodide content was low and there was a large increase in iodide transport into the upper chamber. Cells expressing only PDS accumulated little iodide in the intracellular compartment, but iodide did reach the upper chamber in increased amounts, as compared with wild-type MDCK cells. (From Gillam MP, Sidhaye A, Lee EJ, et al. Functional characterization of pendrin in a polarized cell system. Evidence for pendrin-mediated apical iodide efflux. J Biol Chem 2004;279:13004). |
Functional Characterization of Pendrin
Pendrin is an electroneutral anion exchanger transporting iodide, chloride, bicarbonate, hydroxide, thiocyanate, and formate (83). It remains unclear whether all these anions are physiological ligands. Early functional studies of pendrin in Xenopus oocytes and Sf9 insect cells, as well as studies of cultivated thyrocytes from patients with Pendred’s syndrome revealed that pendrin is unable to transport sulfate despite its homology to sulfate transporters (58,106). In Xenopus oocytes, pendrin mediated uptake of chloride and iodide in a sodium-independent manner (58). The apical localization of pendrin in thyrocytes (72,93), together with its ability to transport iodide in oocytes and the impaired iodide organification found in patients with Pendred’s syndrome (58), suggested a possible role in iodide transport into the follicular lumen (69,72). Functional studies in transfected cells subsequently demonstrated that the protein can indeed mediate iodide efflux (59,60). Evidence for pendrin-mediated apical iodide efflux was obtained in a model system with polarized Madin–Darby canine kidney (MDCK) cells expressing NIS and pendrin (Fig. 4B.4) (61). Consistent with the partial organification defect present in patients with Pendred’s syndrome, naturally occurring mutations of pendrin lead to impaired transport of iodide (59,61,107). Studies in Xenopus oocytes demonstrated that pendrin functions as a coupled, electroneutral iodide/chloride, iodide/bicarbonate, and chloride/bicarbonate exchanger with a 1:1 stoichiometry (108). Iodide was found to be the preferred anion and it is also transported in the presence of high chloride concentrations (108). This latter observation is of particular importance because a physiological role for pendrin-mediated iodide efflux into the follicular lumen is only plausible if the affinity for iodide is higher than for chloride (62,64). Luminal iodide secretion has also been shown in parotid gland ducts, emphasizing that pendrin has a preferential affinity for iodide (108).
It is well established that iodide efflux at the apical membrane is rapidly accelerated by TSH (53,54,55). In cells transfected with pendrin, treatment with forskolin results in a rapid increase in membrane insertion and promotes increased iodide efflux (109), an effect that is mediated by the protein kinase
A pathway based on studies in PCCL3 rat thyroid cells (82). In PCCL3 cells, iodide influx could be inhibited by incubation with an antibody recognizing extracellular conformational epitopes, a finding that supports a role of pendrin in mediating iodide efflux (82). Longer exposure of PCCL3 cells to insulin results in delayed translocation of pendrin from the cytosol to the plasma membrane after activation of the protein kinase C pathway in rat PCCl3 cells (98).
A pathway based on studies in PCCL3 rat thyroid cells (82). In PCCL3 cells, iodide influx could be inhibited by incubation with an antibody recognizing extracellular conformational epitopes, a finding that supports a role of pendrin in mediating iodide efflux (82). Longer exposure of PCCL3 cells to insulin results in delayed translocation of pendrin from the cytosol to the plasma membrane after activation of the protein kinase C pathway in rat PCCl3 cells (98).
In the kidney, pendrin resides on the apical membrane of intercalated type B cells, and in intercalated type non-A-non-B cells in the cortical collecting duct and the connecting tubules (86,87). Type B cells are known to secrete bicarbonate, while type A cells mediate hydrogen secretion (110). Pendrin has been shown to mediate bicarbonate secretion (87), and it contributes to the regulation of blood pressure by modulating renal chloride absorption (111,112,113). In mice, acid-loading results in reduction of the pendrin content in cortical collecting duct cells, and pendrin is relocalized from the apical membrane to the cytosol (114,115). Conversely, bicarbonate loading results in an increase in pendrin in the membranes of these cells (115). Aldosterone and angiotensin II modulate the renal regulation of blood pressure, in part, by regulating pendrin-mediated chloride reabsorption and ENaC-mediated sodium absorption (111,113), thus defining pendrin as a potential target for the treatment of hypertension (112,116). While there is no discernible renal phenotype under basal conditions, severe metabolic alkalosis has been reported in two patients with Pendred’s syndrome confirming that loss of renal bicarbonate excretion by pendrin may lead to life-threatening acid–base disturbances in circumstances of an increased alkali load (117,118).
In the inner ear, pendrin is expressed in specific areas of the endolymphatic system, which is enlarged in patients with Pendred’s syndrome and the Slc26a4-/- mouse (84,85). Functionally, it is essential for maintaining the composition and the potential of the endolymph (119,120,121,122).
Pendred’s Syndrome and its Allelic Variants
Pendred’s syndrome was first recognized by Vaughan Pendred in 1896 (see Chapters 36 and 54) (126). The incidence of Pendred’s syndrome is thought to be as high as 7.5 to 10 in 100,000 individuals, and it is the most common form of syndromic deafness, accounting for about 10% of all patients with hereditary deafness (127,128).
The first insights into the thyroid pathophysiology of Pendred’s syndrome were obtained through the recognition that these patients have a partial defect in the organification of iodide (PIOD) (129). Despite a PIOD, the majority of patients with Pendred’s syndrome are clinically and biochemically euthyroid (130,131). In patients with biallelic mutations in the SLC26A4 gene from countries with a high iodide intake, there are no reports documenting hypothyroidism (132,133,134). On the basis of these observations, it has been suggested that the development of hypothyroidism only occurs under conditions of a low nutritional iodide intake (127,135,136,137,138). Goiter development is highly variable among patients with biallelic mutations in the SLC26A4 gene, even among affected siblings, and in some patients the thyroid is not enlarged and serum thyroglobulin levels are not elevated (65,127). These findings may, in part, be explained by nutritional iodide intake, but most likely this phenotypic heterogeneity also involves the influence of genetic modifiers. The fact that many individuals with biallelic mutations in the SLC26A4 gene have no or only a mild thyroidal phenotype indicates that iodide crosses the apical membrane independently of pendrin through another iodide channel or unspecific channels (62,64). Thus, the relative physiological importance of pendrin in the thyroid needs further characterization. It is also noteworthy, that knockout mice with targeted disruption of the Slc26a4 gene faithfully replicate the phenotype at the level of the inner ear (139,140), but they do not develop a goiter or abnormal thyroid hormone levels (139), even under conditions of iodine deficiency (141,142).
Patients with Pendred’s syndrome from consanguineous families are homozygous for SLC26A4 gene mutations, whereas sporadic cases typically have compound heterozygous mutations (63,83). Mutations in the SLC26A4 gene display impressive allelic heterogeneity, and more than 170 mutations have already been identified (83). Among the mutations characterized in more detail, the mutated pendrin protein is often retained in intracellular compartments, probably the endoplasmic reticulum (ER) (59,61,143). These findings suggest that the mutations result in aberrant folding of the protein, thus preventing full maturation and insertion into the cell membrane. Other mutated pendrin molecules are normally inserted into the membrane, but cannot normally mediate iodide efflux, suggesting that they involve residues that are important for iodide transport (59).
Mutations in the SLC26A4 gene are not only found in patients with classic Pendred’s syndrome, but also in patients with familial enlargement of the vestibular aqueduct (144,145,146). When using a standardized perchlorate test, patients with biallelic mutations in the SLC26A4 gene display a partial iodide organification defect, irrespective of the presence or absence of a goiter, when evaluated by a perchlorate discharge test (146). Others have suggested that a subset of patients with biallelic mutations may be free of any thyroid abnormalities, but the perchlorate test protocol and the threshold used in these studies have not been reported in detail (134,147,148).
Thyroglobulin
Thyroglobulin, a large glycoprotein dimer secreted into the follicular lumen, serves as the matrix for the synthesis of T4 and T, and also as the storage form of the hormones and iodide (see Chapter 5). When T4 and T3 are needed, thyroglobulin reenters the cell to be digested in lysosomes, and T4 and T3 are released into the blood stream (Fig. 4B.1).
 Figure 4B.5 Schematic structure of the thyroglobulin protein with the three repetitive regions, the acetylcholinesterase domain, and the acceptor and donor tyrosyls. |
Structure of the Thyroglobulin Gene and Protein
Thyroglobulin is encoded by a single-copy gene of 270 kb located on human chromosome 8q24.2–8q24.3 (149,150,151,152). It contains 48 exons separated by introns of up to 65 kb
(149,153,154). The promoter region of the gene has remarkable structural similarity with the promoter region of the TPO gene, and is regulated by the transcription factors TTF-1/NKX2.1, TTF-2/FOXE1, and PAX 8 (Fig. 4B.2) (29,30). None of these transcription factors are expressed only in the thyroid, but their combined expression is unique to thyroid follicular cells. The full-length human mRNA contains a 41-nucleotide 5′-untranslated segment preceding an open reading frame of 8,307 bases, and a 3′-untranslated region ranging from 101 to 120 bp (155,156).
(149,153,154). The promoter region of the gene has remarkable structural similarity with the promoter region of the TPO gene, and is regulated by the transcription factors TTF-1/NKX2.1, TTF-2/FOXE1, and PAX 8 (Fig. 4B.2) (29,30). None of these transcription factors are expressed only in the thyroid, but their combined expression is unique to thyroid follicular cells. The full-length human mRNA contains a 41-nucleotide 5′-untranslated segment preceding an open reading frame of 8,307 bases, and a 3′-untranslated region ranging from 101 to 120 bp (155,156).
The thyroglobulin monomer consists of a 19-amino-acid signal peptide followed by 2,749 residues containing 66 tyrosine residues (156). The TG gene contains numerous single nucleotide polymorphisms (SNPs), and generates multiple alternatively spliced transcripts (157). A subset of the SNPs has been associated with a predisposition for autoimmune thyroid disease (158,159). After translation of the mRNA, thyroglobulin monomers are transported to the ER, where they are folded and undergo dimerization. The dimers are then glycosylated. Mature thyroglobulin migrates to the apical membrane in small secretory vesicles and is secreted into the follicular lumen (160,161). In the follicular lumen, thyroglobulin is present as a 19S dimeric glycoprotein of 660 kDa (162).
Analysis of the primary structure of thyroglobulin for internal homology led to its division into four major regions (Fig. 4B.5). The type 1 repetitive region contains 11 segments that contain a cysteine-rich consensus sequence CWCV(D), a sequence found in many proteins (163,164,165). These segments may bind and inhibit cysteine proteases, a feature that could play a role in the processing and degradation of thyroglobulin (see later) (163,164). In the mature protein lacking the signal peptide, these segments are located between amino acids 12 and 1,191 and amino acids 1,492 and 1,546. The type 2 repetitive region, composed of three elements, is located between amino acids 1,437 and 1,484. The type 3 repetitive region is characterized by five elements between residues 1,584 and 2,168. The carboxy-terminal region of the thyroglobulin monomer, encompassing residues 2,192 to 2,716, shares remarkable homology with acetylcholinesterase (165,166). This structure has been interpreted to indicate the possibility of a convergent origin of the TG gene from different ancestral DNA sequences (167). The acetylcholinesterase region is essential for normal conformational maturation, dimerization, and secretion of thyroglobulin (168,169,170).
The maturation of thyroglobulin is controlled by several molecular chaperones such as BiP, GRP94, Erp72 and calnexin (see Chapter 5) (171). Moreover, the acetylcholinesterase domain functions as an intramolecular chaperone (169). Thyroglobulin monomers contain 20 potential glycosylation sites; among them, 16 are known to be glycosylated (172,173), and about 10% of the molecular weight is accounted for by carbohydrates. Other secondary modifications of the protein include sulfation and phosphorylation (174,175). Consensus sequences required for tyrosine sulfation are present at most of the hormonogenic sites within thyroglobulin (176). Tyrosine sulfation may play a role in the hormonogenic process (176).
Mutations in the Thyroglobulin Gene
Recessive mutations in the thyroglobulin gene have been identified in several animal species and humans with goiter and a thyroid function that spans a spectrum from normal function to subclinical or overt hypothyroidism (OMIM# 274700) (see Chapters 36 and 54) (157,177,178,179). The patients typically have very low serum thyroglobulin concentrations (180). Affected individuals are homozygous or compound heterozygous for inactivating mutations in the TG gene (157,178,179,181). Molecular analyses indicate that many of these alterations result in synthesis of thyroglobulin molecules that are retained in the ER and thus fall into the class of ER storage diseases (157,171,182,183).
Thyroid Peroxidase
To serve as an iodinating agent, iodide must be oxidized to a higher oxidation state, a step that is dependent on the presence of H2O2 and is catalyzed by TPO. TPO, a glycoprotein with a prosthetic heme I group, is located at the apical membrane with its catalytic site facing the follicular lumen. In addition to catalyzing the oxidation of iodide, TPO is also essential for the incorporation of iodide onto tyrosine residues in thyroglobulin (organification) and coupling of the iodotyrosines to generate T4 and T3 (Fig. 4B.1).
The purification and the biochemical properties of TPO were discussed comprehensively in previous versions of this chapter by A. Taurog (184,185). The role of TPO as an autoantigen in autoimmune thyroid disease is discussed in Chapter 35 (186).
The Gene and Protein Structure of Thyroid Peroxidase
The human TPO gene is located on chromosome 2pter-p12 (187,188), spans about 150 kb, and consists of 17 exons separated by 16 introns (189). The first cDNAs encoding TPO were isolated from human and porcine cDNA libraries (186,187,190,191). The full-length human TPO cDNA encodes a protein of 933 amino acids (187). In addition to the full-length mRNA encoding full-length TPO (TPO1), several shorter transcripts of unknown biologic importance have been identified (187,192,193). The most abundant alternative
transcript lacks 171 nucleotides, secondary to deletion of exon 10, which encompasses codons 533–590 (TPO2) (187,194). The encoded protein lacks enzymatic activity (194). In immunoblot analyses, TPO appears as a doublet of 110 and 105 kDa, a phenomenon that is not explained by translation of TPO2 (195).
transcript lacks 171 nucleotides, secondary to deletion of exon 10, which encompasses codons 533–590 (TPO2) (187,194). The encoded protein lacks enzymatic activity (194). In immunoblot analyses, TPO appears as a doublet of 110 and 105 kDa, a phenomenon that is not explained by translation of TPO2 (195).
The amino terminus of TPO is located in the lumen of thyroid follicles, and the extracellular domain forms a loop created by two (human) or one (porcine) intramolecular disulfide bonds, which is followed by a single membrane-spanning domain in close proximity to its carboxy terminus (196,197). Introduction of a stop codon immediately upstream of the putative transmembrane domain (amino acids 846 to 870) results in a soluble protein that is secreted into the medium and retains enzymatic activity (198). Human TPO has five potential glycosylation sites, and about 10% of its weight is carbohydrate (185). The location and nature of the N-linked oligosaccharide units have been determined only for porcine TPO (199).
The prosthetic heme group, a bis-hydroxylated heme I that is distinct from the heme b (protoporphyrin IX) found in many other hemoproteins, is covalently bound to glutamine 399 and aspartate 238 of the apoprotein (200). Amino acid side chains directly coordinated to the iron atom or positioned in its immediate proximity are critical for modulating enzyme reactivity and specificity. This involves two histidine residues located on opposite sides of the heme moiety, a proximal histidine coordinately linked to the iron center, and a distal histidine located in proximity of the peroxide-binding pocket (200,201,202). Based on sequence alignment with myeloperoxidase (MPO), the distal histidine in TPO is located at position 239, and the proximal histidine at position 494 (200,202). The distal histidine and a nearby arginine residue at position 396 are thought to be involved in the formation of compound I, a two-electron oxidized form of TPO (see later) (200,202). TPO2 is enzymatically inactive (194), presumably because it lacks asparagine 579, which forms a stabilizing hydrogen bond with the proximal histidine 494 (200,202).
All mammalian peroxidases belong to the same gene family (202). TPO has a high degree of sequence similarity with MPO (190,203,204,205) and other mammalian peroxidases (206,207,208). The first 735 amino acids of TPO have 42% sequence identity with MPO (191), and the homology between TPO and MPO in the vicinity of the heme group is as high as 74% (190). Based on sequence analyses, which have revealed domains homologous to epidermal growth factor, complement C4b, and mitochondrial cytochrome C oxidase I, the TPO gene may be a mosaic gene (191). The structure of MPO has been solved at 3 Å resolution (209), and more recently at a 1.8 Å resolution (210,211). Theoretical three-dimensional structures have been proposed for other mammalian peroxidases using the MPO model as a scaffold (201), but the three-dimensional structure of TPO has not been modeled (185). TPO, analogous to MPO (209), may form a disulfide-linked dimer in its native form, based on immunoblot experiments under non-reducing conditions (212,213), a model that has been questioned (195).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


