Mature T-cell and NK-cell neoplasms
Mature B-Cell neoplasms
Mycosis fungoides
Cutaneous marginal zone B-cell lymphoma (MALT type)
Pagetoid reticulosis (localised disease)
Cutaneous follicle centre lymphoma
Follicular, syringotropic, granulomatous variants
Cutaneous diffuse large B-cell lymphoma
Granulomatous slack skin
Intravascular large B-cell lymphoma
Sezary syndrome
Lymphomatoid granulomatosis
CD30+ T-cell lymphoproliferative disorders of the skin
Chronic lymphocytic leukaemia
Lymphomatoid papulosis
Mantle cell lymphoma
Primary cutaneous anaplastic large-cell lymphoma
Burkitt lymphoma
Subcutaneous panniculitis-like T-cell lymphoma
Primary cutaneous peripheral T-cell lymphoma (PTL), unspecified
Immature haematopoietic malignancies
(Subtypes of PTL, provisional)
Primary cutaneous aggressive epidermotropic
Blastic NK-cell lymphoma
CD8-positive cytotoxic T-cell lymphoma
CD4+/CD56+ haematodermic neoplasm
Cutaneous gamma-/delta-positive T-cell lymphoma
Precursor lymphoblastic leukaemia/lymphoma
Primary cutaneous small/medium CD4+ T-cell lymphoma
T-lymphoblastic leukaemia
Extranodal NK/T-cell lymphoma, nasal type
T-lymphoblastic lymphoma
Hydroa vacciniformia-like lymphoma (variant)
B-lymphoblastic leukaemia
Adult T-cell leukaemia/lymphoma
B-lymphoblastic lymphoma
Angioimmunoblastic T-cell lymphoma
Myeloid and monocytic leukaemias
Hodgkin lymphoma
Mature T-Cell and NK-Cell Neoplasms
Mycosis Fungoides and Subtypes
Clinical features: Mycosis fungoides (MF) represents the most common type of cutaneous T-cell lymphomas. It has been classified according to the type of skin lesions (patches, plaques and tumours), the presence or absence of large-cell transformation and/or extracutaneous involvement. It pursues an indolent clinical course with slow progression, and sometimes, subsequent biopsies are needed for a proper diagnosis, especially in the early manifestations of the disease. The male to female ratio is 2:1 with adults/elderly mostly affected. Skin lesions vary from large erythematous patches preferentially involving non-sun-exposed areas to reddish brown infiltrated plaques with wrinkled surface, to nodules solitary or generalised, sometimes ulcerated. A combination of patches, plaques and tumours is common in the well-developed disease. Large-cell transformation occurs in more than 50 % of tumour stage MF. The most frequent sites of extracutaneous involvement in the later stages of the disease are lymph nodes, lung, spleen and liver. Several clinical variants of MF have been described. Some of them represent distinct clinico-pathological entities, while others are peculiar kind of skin involvement described by case reports. MF is rare in childhood (0.5–5 % of all cases) and presents mainly with early-stage lesions sometimes with concurrent or prior history of pityriasis lichenoides chronica. Sezary syndrome (SS) is actually considered an aggressive form of peripheral T-cell lymphoma involving elderly adults which may be or not preceded by idiopathic erythroderma and presents with generalised lymphadenopathy and pruritic erythroderma. The disorder is characterised by the presence of the so-called Sezary cell in the peripheral blood, the lymph nodes and the skin.
Histopathology: The histological features of MF are prototypical and correlate with the different clinical lesions. Patch stage: The diagnostic architectural hallmarks consist in the so-called string of pearls (at least four T-lymphocytes contiguously aligned within the basal layer), associated with Pautrier microabscesses (collection of at least four atypical T cells within the epidermis and papillary dermal fibrosis, with coarse horizontally disposed wiry bundles of collagen). MF cells (Lutzner cells) are lymphocytes of medium-large size (approximately the diameter of a basal keratinocyte nucleoli), with irregular convoluted nuclei (cerebriform nuclei) (Fig. 9.1a and b). These epidermotropic T cells are larger than those present in the papillary dermis and show a discrete halo. Often in Pautrier microabscesses they are intermingled with dendritic Langerhans cells. Absence or little spongiosis is mandatory. A clinico-pathological algorithm has been published [10] reporting different parameters to make diagnosis most reproducible. Plaques stages: The neoplastic cells represent the main contributor to dermal infiltrate unlikely patch stage. Epidermotropism is prominent sometimes associated with syringotropism and Pautrier microabscesses most frequent (Fig. 9.2). The tumour cells are highly atypical and form a dense band-like infiltrate within the papillary dermis and perivascular aggregates in the middle dermis. Tumour stage: A pan-dermal dense neoplastic infiltrate is typical of this stage with extension to the subcutaneous fat and possible sparing of the epidermis. Large-cell transformation: It is defined by the presence of more than 25 % large neoplastic cells forming clusters.
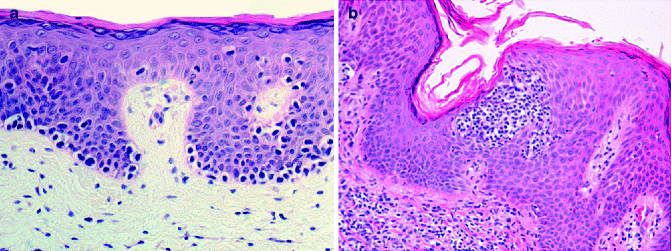
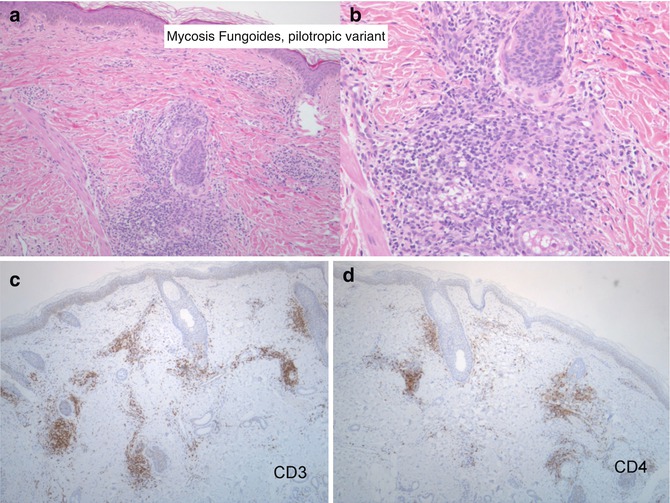
Fig. 9.1
Mycosis fungoides, patch stage. (a) Atypical lymphocytes arranged along the interface line. (b) Intraepidermal collection of atypical cells forming Pautrier microabscess
Fig. 9.2
Mycosis fungoides, pilotropic variant. (a, b) Morphologic detail of the relationship between the lymphoid infiltrate ad the pilo-sebaceous unit. (c, d) The lymphoid cells express the CD3 and CD4 antigen
Immunophenotype: MF cell typically expresses CD2, CD3, CD4 and CD5. The diagnosis of MF is most challenging for the pathologist in the early stages because most dermal lymphocytes are reactive. Plenty of studies have tried to determine reproducible cut-offs of the CD8-/CD3-positive cells in order to help discriminating early MF from reactive dermatoses. Assessing of CD4 cells has been avoided because of cross-reaction with histiocyte population. Different studies show that CD8/CD3 is significantly lower in MF when compared with controls, and other reports suggest that CD4/CD8 more than 2 is specific for MF. Actually semiquantification of lymphocyte subsets is unlikely to be routinely used. On the contrary, of more importance is the detection of T-cell-associated antigen loss, with special regards to CD7 and at less extent CD5 downregulation. The suggested immunohistochemical baseline includes a pan-B-cell marker (CD79a). With disease progression CD30 expression may be detectable in the large-cell component. Neoplastic cells of erythrodermic MF and SS share the same immune signature, but CD7 antigen loss is less frequent in the latter entity. Moreover, large cells in SS are often MUM1 positive.
Genetics: T-cell receptor genes are clonally rearranged in most of the cases of MF in plaques or tumour stage, while only about 50 % of the patch stages do not show any TCR gene rearrangement. Complex karyotypes are present in tumour stages of the disorders with many structural and numerical alterations described.
Primary Cutaneous CD30-Positive Lymphoproliferative Disorders
CD30 T-cell lymphoproliferative disorders comprise a spectrum of conditions ranging from lymphomatoid papulosis (Lyp) to anaplastic large-cell lymphoma (ALCL) and including borderline cases [21]. This group accounts for 30 % approximately of cutaneous T-cell lymphomas and has to be distinguished by cases of transformed mycosis fungoides expressing CD30 antigen (a member of the tumour necrosis factor receptor family).
Extensive CD30 expression by the neoplastic population is mandatory for diagnosis of these entities which share a common pathogenetic pathway strictly involving the role of CD30/CD30L in activation of TRF2 (TNF receptor- associated factor) up to NF-kB signal transduction leading either to proliferation of apoptosis.
Owing to this mechanism, these lymphoproliferative disorders run a more favourable course and are amenable of regression. Most probably, past diagnosis of “regressing atypical histiocytosis” and “cutaneous Hodgkin disease” would be nowadays renamed as PC CD30 LPD.
It is of the utmost relevance to keep in mind that CD30-positive lymphocytes isolated or in clusters can be observed in a variety of inflammatory skin conditions, such as viral infections, arthropod bite reactions, lymphomatoid drug reaction and lupus pernio [22].
Lymphomatoid Papulosis (Lyp)
Clinical features: Red papules and nodules smaller than 1.5–2 cm or pink papulo-nodules, single or in clusters, predominantly affecting the trunk and limbs mostly of adults but even of children. The lesion may ulcerate in the centre and result in a scar. A waxing and waning course of the eruption is typical with tendency to spontaneous involution in 3–4 months and subsequent recurrences. The incidence of concurrent lymphoma or progression to lymphoma ranges between 4 and 20 % confirming the benign clinical course of this disorder. Yet, other malignant skin lymphomas, in particular mycosis fungoides, anaplastic large-cell lymphoma and Hodgkin lymphoma, may precede or follow Lyp.
Histopathology: Three main histologic subtypes have been described, with features merging one in the other and varying in relation to the age of the biopsied lesion.
Type A Lyp presents as a bottom-heavy striking polymorphous infiltrate with a distinct perivascular, perineural and eccrinotropic pattern, composed of small reactive lymphocytes, histiocytes and rare Sternberg-like atypical cells. An epidermal reaction with neutrophilic exocytosis and even necrosis and ulceration sustained by heavy vasculitis is frequent. Sometimes a granulomatous component with eosinophilia is evident so mimicking arthropod bite reaction. In type B Lyp, which is the less frequent variant, the infiltrate is still mixed, but large atypical cells begin to dominate and show a lichenoid distribution, resembling the plaque stage of mycosis fungoides. Type C Lyp lesions are featured by monotonous infiltrates of the large atypical cells and few small lymphocytes intermingled.
Immunophenotype: The large atypical cells of types A and C are CD30 positive and usually CD4+, so sharing the same signature of ALCL. In contrast, the large cells of Lyp B do not express CD30. Markers pointing to natural killer differentiation are generally negative, while cytotoxic antigen can be variably expressed.
A rare CD8+ form of Lyp has been described, histologically characterised by a granulomatous eccrinotropic infiltrate.
Genetics: t(2;5) translocation responsible of the protein ALK (anaplastic lymphoma kinase) has been occasionally reported. On the contrary T-cell gene rearrangement has been described in up to 60–70 % cases of Lyp.
Cutaneous Anaplastic Large-Cell Lymphoma
Clinical features: Usually single, but sometimes multiple, persistent large often ulcerative nodule, which does not undergo spontaneous regression. A cut-off of 2 cm in diameter has been proposed by some investigators to distinguish from Lyp. The median age of presentation is 60 years. A type C Lyp/borderline CD30-positive lymphoproliferative disease has been described, usually associated with a history of Lyp either concurrent or prior.
Secondary skin involvement of primary nodal ALCL, occurrence of ALCL in the setting of other lymphomas (mycosis fungoides and Hodgkin lymphoma) and ALCL in the context of post-transplant T-cell lymphoproliferative disorders represent other categories of this disease.
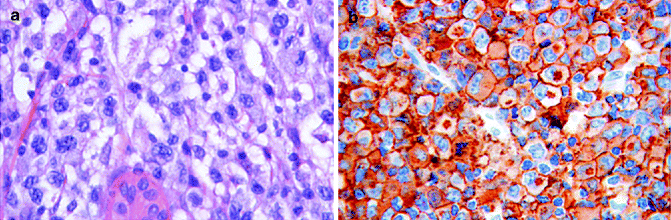
Histopathology: The common pattern shows a cohesive sheet-like growth, extending to the subcutis, of large cells, even multinucleated, with eccentric horseshoe-shaped nucleus and prominent eosinophilic Golgi region (Fig. 9.3). A small-cell variant exists with scattered atypical hallmark cells intermingled with dominant small-intermediate-sized atypical lymphocytes. Angiocentricity and angiodestruction by the neoplastic cells are frequent findings. Epidermotropism is reported. The epitheliomorphic feature of the cells can represent a challenging problem of differential diagnosis on morphological grounds with undifferentiated carcinoma. Other histological subtypes are the sarcomatoid, giant, neutrophilic-rich and histiocyte-rich variants. Mixed patterns are frequent. Histological variant does not influence prognosis with the exception of small-cell subtype, which runs a worse clinical course.