Myeloproliferative Neoplasms
POLYCYTHEMIA VERA
 INTRODUCTION
INTRODUCTION
Polycythemia vera (PV) is a clonal disorder of a multipotent hematopoietic stem cell in which overproduction of morphologically normal red cells, white cells, and platelets occurs in the absence of an apparent cause. The commonest of the chronic myeloproliferative disorders, PV, is uncommon, occurring at an average frequency of 2/100,000, but with increasing age, rates as high as 18/100,000 have been observed. Females predominate, particularly below age 40 years.
 PATHOGENESIS
PATHOGENESIS
The etiology of PV is unknown. Abnormalities of chromosomes 1, 8, 9, 13, and 20 have been identified in up to 30% of PV patients, but they are neither specific for the disorder nor necessary for its pathogenesis; in many instances they appear to occur as secondary events and their expression can be enhanced by exposure to chemotherapeutic agents (1). Erythropoietin-independent in vitro erythroid colony formation is a characteristic feature of PV, although not specific for it, since this behavior has also been observed in primary myelofibrosis (PMF) and essential thrombocytosis. Constitutive activation of JAK2 (2), which is the cognate tyrosine kinase for type 1 hematopoietic growth factor receptors such as the erythropoietin, thrombopoietin, and granulocyte colony-stimulating factor receptors, has been identified to be the molecular basis for such growth factor independence in PV and its companion myeloproliferative disorders PMF and essential thrombocytosis.
The mechanism for constitutive JAK2 activation in the chronic myeloproliferative disorders is an acquired point mutation in the autoinhibitory JH2 domain of the JAK2 gene, replacing valine with phenylalanine (V617F). JAK2 is located on the short arm of chromosome 9 and loss of heterozygosity for 9p is a common cytogenetic lesion in PV, leading to homozygosity for the JAK2 V617F mutation; in some patients, there is also reduplication of chromosome 9. In PV, approximately 90% of patients express the JAK2 V617F mutation, of which approximately 35% are homozygous for it. Approximately 5% have a JAK2 exon 12 activating mutation. No clinical differences have been identified between heterozygotes and those homozygous for JAK2 V617F, nor are there any clinical differences between PV patients expressing JAK2 V617F and those who do not. Thus, although the JAK2 V617F mutation provides an explanation for the hematopoietic growth factor independence of PV hematopoietic cells in vitro, their apoptosis resistance and their uncontrolled growth in vivo, the absence of the mutation in some patients with classical PV, and its expression in PMF and essential thrombocytosis patients strongly suggest that other as yet unidentified molecular lesions are involved in the pathogenesis of these disorders.
 CLINICAL FEATURES
CLINICAL FEATURES
PV is extremely variable in its presenting manifestations as well as its clinical features, which also change over the course of the disorder. Because its onset can be insidious, an abnormal blood count is often the first sign of the disease. In approximately 40% of patients, there will be an increase in red cells, white cells, and platelets. In approximately 15% of patients, erythrocytosis will be the sole presenting manifestation. In approximately 5%–10% of patients, an elevated platelet count may be the first manifestation of the disease, while in the rest, erythrocytosis and thrombocytosis or leukocytosis are the presenting blood abnormalities. Extramedullary hematopoiesis as manifested by palpable splenomegaly occurs in approximately 40% of patients at the time of diagnosis; rarely, myelofibrosis can be the initial manifestation of PV with erythrocytosis becoming evident later on. Since PV is a hypercoagulable state, arterial or venous thrombosis may also be the first manifestation of the disease. Classically, in young women, the thrombosis most commonly involves the hepatic veins, often as the presenting manifestation and often with an apparently normal hematocrit due to concomitant plasma volume expansion. Pruritus, usually aquagenic, is also not uncommon as a presenting manifestation, but PV is often not initially recognized as its cause. Erythromelalgia, in which the extremities become warm, red, and painful; migraine headaches; or other neurologic disturbances such as vertigo or visual disturbances are also characteristic symptoms that indicate the presence of an elevated red cell mass or thrombocytosis.
 LABORATORY ABNORMALITIES
LABORATORY ABNORMALITIES
In addition to increases in the red cell, granulocyte, and platelet counts, the MCV can be low if red cell mass expansion or gastrointestinal blood loss depletes body iron stores. An elevated leukocyte alkaline phosphatase and serum vitamin B12 and vitamin B12 binding capacity due to increased release of granulocyte transcobalamin III reflect neutrophil activation, presumably due to JAK2 V617F, which is also responsible for the increased expression of granulocyte PRV-1 mRNA (CD177). When the platelet or leukocyte counts are elevated, spurious hyperkalemia may be observed, as can hypoglycemia and a low pO2 if blood samples are not collected on ice and in the presence of sodium azide. Elevation of the serum alkaline phosphatase occurs with extramedullary hematopoiesis and becomes more marked after splenectomy.
Abnormalities of coagulation in PV are largely limited to platelet function. These include defective platelet aggregation to ADP, epinephrine, or collagen alone or in combination and loss of alpha granules and dense bodies. When the platelet count exceeds 1,000,000/ml, higher molecular weight von Willebrand multimers will be absorbed by the platelets and degraded, leading to a reduction in ristocetin cofactor activity and an acquired form of von Willebrand’s disease, although spontaneous bleeding due to this is uncommon.
 DIAGNOSIS
DIAGNOSIS
Elevation of the red cell mass is the sine qua non of PV, the only feature that distinguishes it from its companion myeloproliferative disorders, PMF and essential thrombocytosis, and the feature of the disease that is responsible for its most frequent serious consequences, thrombosis and hemorrhage. Unfortunately, erythrocytosis is not unique to PV, and in recent years the very means for identifying the presence of erythrocytosis, direct determination of the red cell mass by isotope dilution, has become unavailable in many medical centers. Attempts to resolve this issue by the use of surrogate markers for direct red cell mass determination have not proved to be useful (3). For example, specific hematocrit or hemoglobin levels are woefully inadequate as indicators of the red cell mass unless the hematocrit is >60% (hemoglobin >20 g/dl) in a man, or >52% in a woman (hemoglobin >17 g/dl). The reasons for this are a consequence of blood rheology and the unique pathophysiology of PV with respect to blood volume regulation.
For example, when erythrocytosis occurs as a consequence of hypoxia, there is a simultaneous reduction in the plasma volume as the body attempts to maintain a normal total blood volume. This contributes to the observed increase in hematocrit. In PV, however, particularly in women, as the red cell mass rises, the plasma volume either rises or fails to decrease. Furthermore, with splenomegaly, there is a compensatory increase in plasma volume. Both of these situations lead to hematocrit values that are spuriously low with respect to the actual red cell mass (4). As a corollary, a decrease in the plasma volume alone can lead to a falsely elevated hematocrit, when in fact the red cell mass is normal.
The recent discovery of the JAK2 V617F mutation has greatly simplified the evaluation of a high hematocrit and the diagnosis of PV. This is because first, benign disorders causing erythrocytosis are more common than PV (Table 26-1) and second, because surrogate markers for the latter lack sensitivity and specificity. For example, while the serum erythropoietin level is lower in PV than in other disorders causing erythrocytosis, the serum erythropoietin level can also be normal in PV as well as in secondary forms of erythrocytosis. Similarly, the bone marrow examination can be normal in PV or even mimic that of PMF or essential thrombocytosis. Cytogenetic abnormalities are present in only 30% of PV patients and are not pathognomonic for the disease, while other markers such as elevation of the leukocyte alkaline phosphatase and endogenous erythroid colony formation are merely consequences of the constitutively active JAK2.

Figure 26-1 illustrates an algorithm for the evaluation of the patient with a high hematocrit. When red cell mass and plasma volume determinations are not available, it is reasonable to start with an assay for JAK2 V617F with the knowledge that a positive assay only indicates the presence of a myeloproliferative disorder, while a negative assay does not exclude such a disorder. In the absence of a red cell mass determination, a positive JAK2 V617F assay in a patient with a high hematocrit obligates the physician to phlebotomize the patient to the normal hematocrit for gender as discussed below.
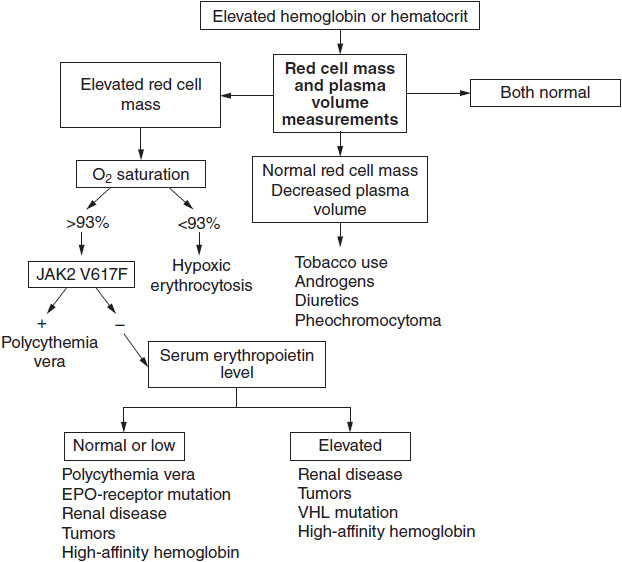
FIGURE 26-1 Algorithm for the diagnosis of polycythemia vera. The first requirement is to establish the basis for an elevated hemoglobin or hematocrit. If it is determined that there is an elevated red cell mass, an assay for JAK2 V617F will establish the diagnosis in over 90% of patients with polycythemia vera. A negative JAK2 V617F assay does not, however, exclude a myeloproliferative etiology and in the absence of splenomegaly, leukocytosis, or thrombocytosis; further studies will be required.
 NATURAL HISTORY
NATURAL HISTORY
Most classical hematology textbooks suggest that the natural history of PV follows an inevitable course from erythrocytosis through myelofibrosis and myeloid metaplasia to acute leukemia if the patient does not die first from some other complication or comorbidity. This depiction ignores the clinical heterogeneity of the disease, its modification by improved therapies, and the earlier stages at which PV is now usually recognized. In this regard, prognosis does not appear to be influenced by the presence or the absence of JAK2 V617F or whether this mutation is expressed homozygously or heterozygously.
The complications of PV are listed in Table 26-2. Erythrocytosis, not thrombocytosis, is responsible for the major thrombotic complications of PV; minor transient thrombotic or ischemic complications such as erythromelalgia, ocular migraine, or digital infarction do involve the platelets but are exacerbated by erythrocytosis, which promotes platelet activation, platelet- leukocyte interactions, as well as endothelial cell activation and damage, all of which enhance thrombogenesis. Erythrocytosis can also cause hypertension, splenomegaly, and exacerbate aquagenic pruritus. Acid-peptic disease leading to gastrointestinal hemorrhage and iron deficiency occur at a higher frequency in PV patients than in the general population; the roles of vascular stasis, excess histamine, or other cytokine production are unknown, but the frequency of Helicobacter infection is increased in PV.
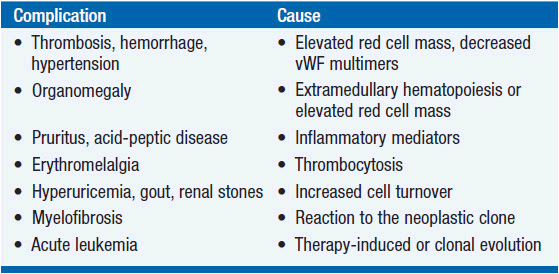
Over time, there will be a gradual increase in the leukocyte and platelet counts, but the leukocytosis is not usually progressive unless there is disease acceleration, while asymptomatic thrombocytosis requires no therapy. The development of excessive extramedullary hematopoiesis with massive splenomegaly and hepatomegaly is a serious complication of PV, occurring in about 10%–15% of patients. These patients are at a higher risk of subsequent leukemic transformation (5).
Splenomegaly can lead to mechanical discomfort, easy satiety, portal hypertension, and cachexia. Marrow fibrosis is another expected event in the natural history of PV. It is essential to distinguish between the development of increased marrow reticulin as a consequence of marrow cell hyperplasia and the hematopoietic stem cell disorder, PMF. There is no evidence that myelofibrosis in PV represents a bad prognostic sign or that it impairs marrow function in the absence of exposure to agents that damage the bone marrow; it is stem cell failure that is the problem. Rarely, pulmonary hypertension has developed with long-standing disease; in some patients this may be due to extramedullary hematopoiesis, while in others there may be pulmonary fibrosis.
Spontaneous acute leukemia develops in PV at an incidence of approximately 1.5%–2.5%; this usually occurs within the first 8 years of the disease and most commonly in patients older than 60 years. Chemotherapy or radiation-induced acute leukemia occurs at rates as high as 10% when these patients are exposed to 32P or alkylating agents. The role of hydroxyurea as a leukemogen has been a matter of debate, but in one randomized prospective clinical trial (6, 7), hydroxyurea was associated with a 10% incidence of acute leukemia after 10 years; hydroxyurea is also a proven tumor promoter when used in conjunction with 32P or an alkylating agent or with UV light exposure.
 TREATMENT
TREATMENT
PV is generally an indolent disease in which survival is measured in decades in the majority of patients. Most estimates of disease survival have failed to take into account the toxic forms of therapy that have been generally employed, the inadequate use of phlebotomy, and the later stages at which the disease was previously recognized clinically. Furthermore, it is now apparent that PV is a heterogenous disorder with both indolent and aggressive forms and that aggressive chemotherapy has not improved survival (8). There is currently no curative therapy for PV with the possible exception of allogeneic bone marrow transplantation, a therapy not suitable for the older patients who most commonly develop this disorder (9). Thus, treatment should be tailored to disease manifestations. Unfortunately, in contrast to PMF, prognostic risk stratification according to laboratory features has not yet been possible with the exception that a prior history of thrombosis is an adverse risk factor for recurrent thrombotic events.
Erythrocytosis is the greatest initial threat to health because of the adverse effects of hyperviscosity (thrombosis, hemorrhage, hypertension, headache, and impaired cognitive function). Therefore, the red cell mass should be lowered by phlebotomy to achieve a hematocrit of ≤42% (hemoglobin ≤12 g%) in women and ≤45% (hemoglobin ≤14 g%) in men (8). This can be done quickly in all but the frailest because phlebotomy stimulates rapid plasma volume expansion. Repeated phlebotomies will be necessary to maintain the hematocrit at a normal level and to induce iron deficiency, but once this is achieved, the need for phlebotomy will diminish. Phlebotomy therapy actually improves platelet function, does not contribute significantly to thrombocytosis, and does not lead to myelofibrosis, and it must be remembered that the higher the hematocrit, the greater the extent of tissue damage with thrombosis (8). Pruritus, usually aquagenic, is a distressing symptom in approximately 30% of patients. There is no single effective remedy. Phlebotomy, antihistamines, PUVA light therapy, interferon alpha, and hydroxyurea have all been effective but none uniformly. Hyperuricemia (uric acid >10 mg/dL) responds well to allopurinol.
Platelet-related microvascular complications include migraine, visual auras, transient ischemic attacks, erythromelalgia, and digital infarction. Aspirin is a specific remedy for erythromelalgia but with migraine, it may be necessary to lower the platelet count as well to achieve relief using conventional remedies. Symptomatic thrombocytosis causing acquired von Willebrand’s disease will also require platelet count reduction. Asymptomatic thrombocytosis without a significant reduction in ristocetin cofactor activity (<30%) requires no treatment in the absence of a thrombotic risk factor. In this regard, it is important to emphasize that there is no correlation between the platelet count and thrombosis, and no study to date has demonstrated that in the absence of hematocrit control, platelet count reduction prevents arterial or venous thrombosis. Hydroxyurea does appear to be more effective than anagrelide in the prevention of transient ischemic attacks but not venous or arterial thrombosis. The use of prophylactic low dose aspirin therapy is no substitute for adequate control of the red cell mass and has not been demonstrated to have clinical efficacy in asymptomatic PV patients who are adequately phlebotomized.
Control of extramedullary hematopoiesis involving the spleen and liver is the most challenging therapeutic problem in PV but fortunately not one that involves every patient. Interferon alpha, and its pegylated congener in particular, is the drug of choice for this because it lacks the potential for bone marrow damage (10). A recent study demonstrated that durable molecular remissions could be achieved with pegylated interferon (11). Since interferon’s side effects can be significant with chronic use, in the absence of a complete molecular remission, intermittent use is a prudent strategy. In some patients, splenomegaly may be refractory to interferon and chemotherapy, and mechanical discomfort, cachexia, and portal hypertension will demand treatment. If bone marrow transplantation is not an option, the newly approved nonspecific JAK2 inhibitor, ruxolitinib, is the drug of choice in this situation (see the primary myelofibrosis chapter). Low-dose thalidomide is another option worth considering with surgery as the choice of last resort because of the high complication rate associated with it. The postoperative complications of splenectomy include wound dehiscence, hernias, bleeding, portal or mesenteric vein thrombosis, exuberant hepatic extramedullary hematopoiesis, and extreme leukocytosis and thrombocytosis, all of which can be very difficult to control. Splenic irradiation is only a temporary solution and not advisable unless surgery is not an option (12).
 PREGNANCY
PREGNANCY
The opportunity for pregnancy should not be denied to women with PV who have no medical contraindications and prior thrombosis is not one of these. The major threat to a successful outcome is failure to maintain the red cell mass at a safe level. Since there is an expansion of the plasma volume with pregnancy normally, there will be masking of the expanded red cell mass. A normal hematocrit in a pregnant woman is never normal and this is doubly true in PV. It is essential to phlebotomize these patients to a hematocrit of <33% and avoid iron supplements; folic acid supplementation is mandatory. Thrombocytosis and splenomegaly may mandate the use of interferon alpha. Given the elevation of von Willebrand factor that occurs during pregnancy, aspirin therapy may be prudent but this is unproved.
PRIMARY MYELOFIBROSIS
 INTRODUCTION
INTRODUCTION
Primary myelofibrosis (PMF) is the least common and most enigmatic of the chronic myeloproliferative disorders. Most frequent after age 60 years, PMF has an incidence of approximately 1/100,000 with male predominance. Previously known as agnogenic myeloid metaplasia, idiopathic myelofibrosis, primary osteomyelofibrosis, or myelofibrosis with myeloid metaplasia, it is important to note that both the first and last appellations actually describe a pathologic process that is not restricted to the disease PMF but can be caused by a variety of benign and malignant processes (Table 26-3). Like its companion myeloproliferative disorders, PV and essential thrombocytosis, PMF is a clonal hematopoietic stem cell disorder, but in contrast to them, it is associated not only with overproduction of blood cells without an obvious cause but also, in many patients, with anemia, leucopenia, or thrombocytopenia.
 PATHOGENESIS
PATHOGENESIS
The etiology of PMF is unknown. Although irradiation and exposure to organic chemicals such as toluene and benzene can cause marrow fibrosis, no other consistent environmental risk factors have been identified for PMF and familial transmission is rare. Cytogenetic abnormalities occur in more than 50% of patients but generally involve the same chromosomes as in PV and essential thrombocytosis, and none appear to be involved in its pathogenesis. Myelofibrosis is the hallmark of the disorder, but there is good retrospective histologic evidence that a premyelofibrotic phase of the disease exists (13), supporting other evidence that the fibrosis is a consequence of the disease, not its cause.
TABLE 26-3 DISORDERS CAUSING MYELOFIBROSIS
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


