Molecular Targeted Drugs
INTRODUCTION
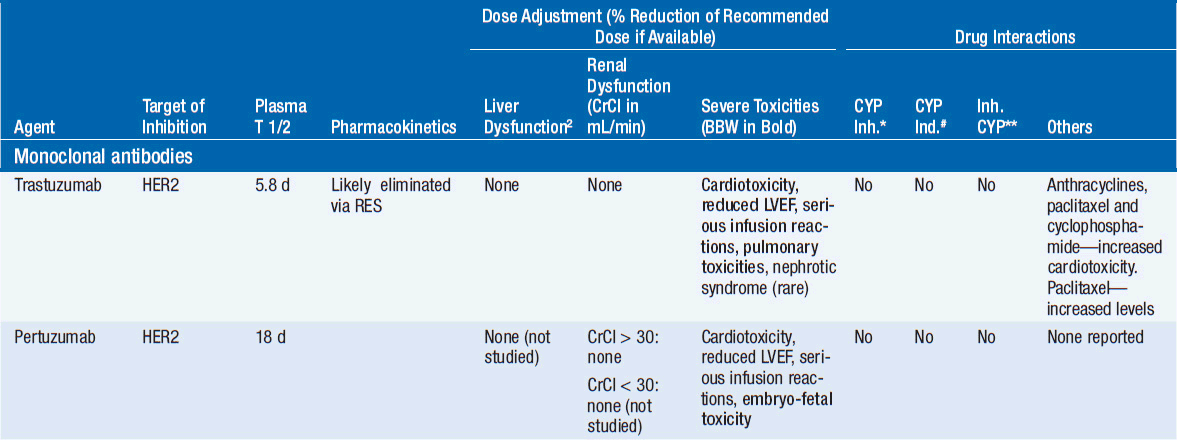
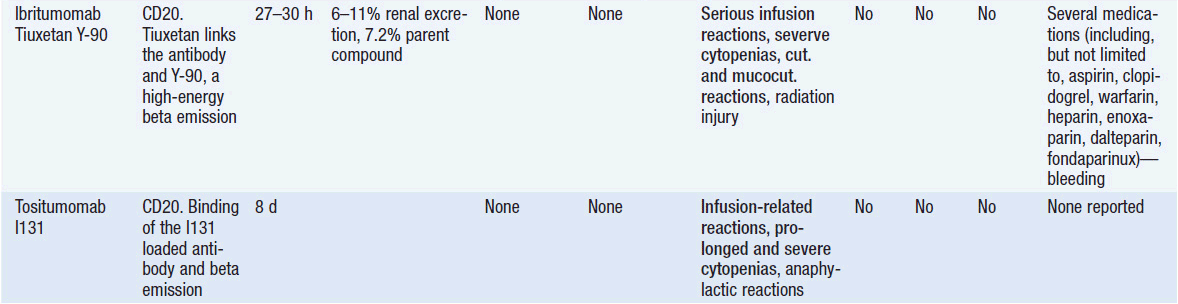
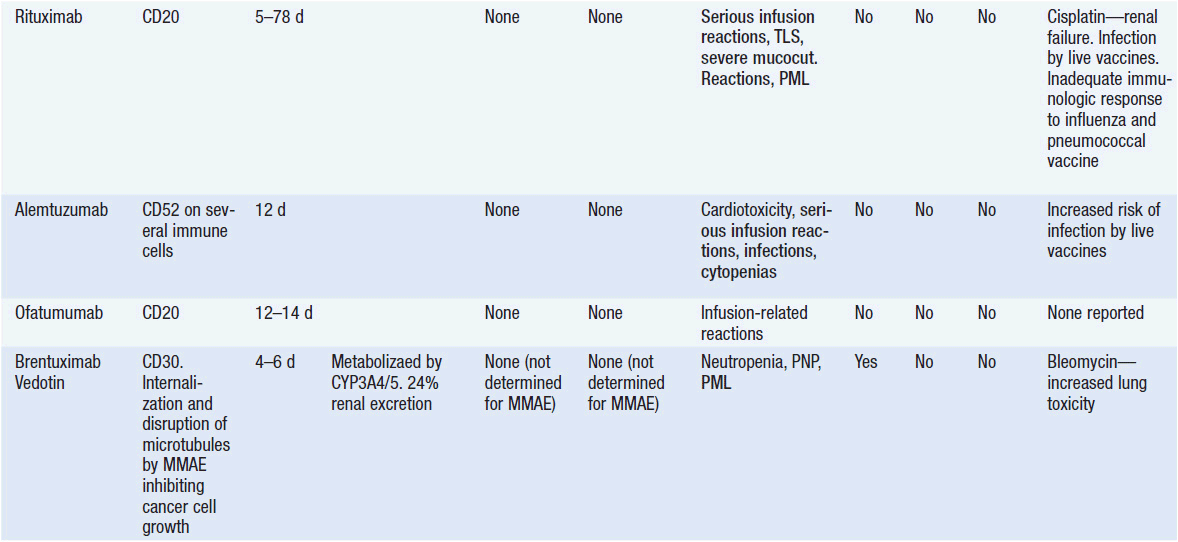
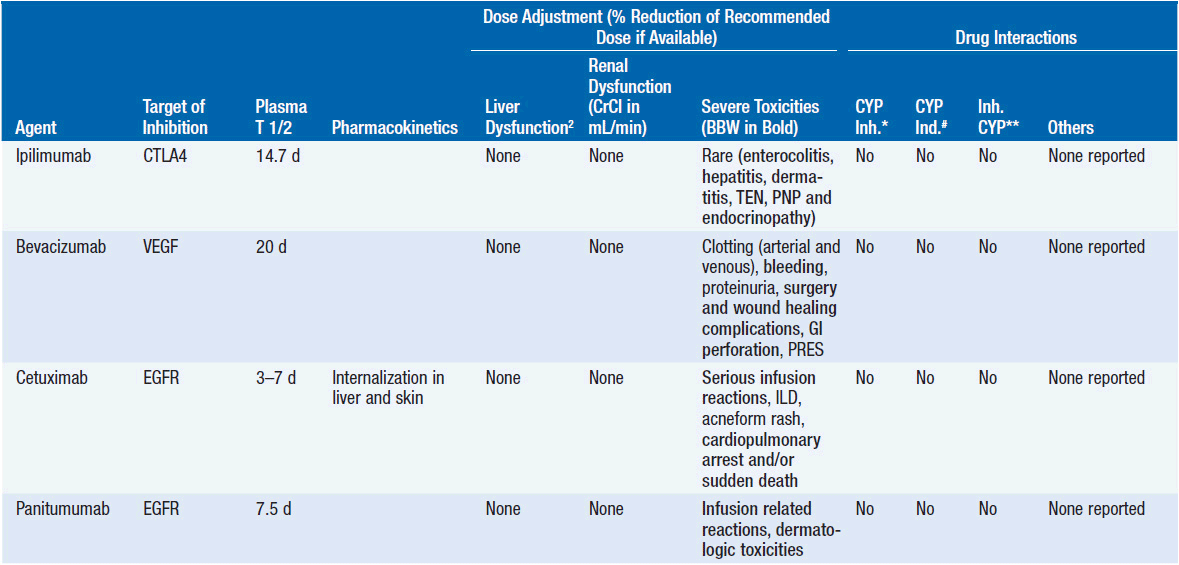
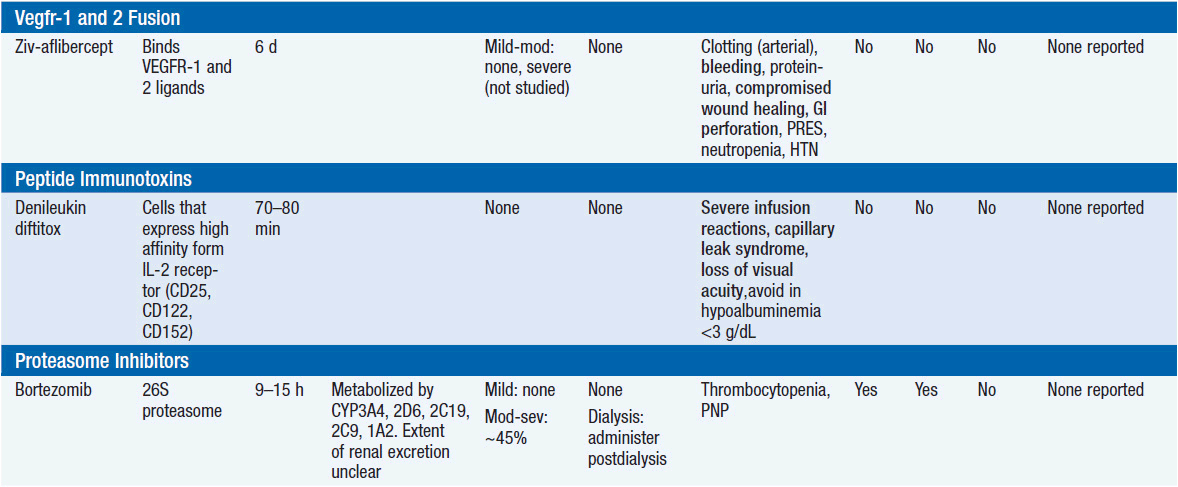
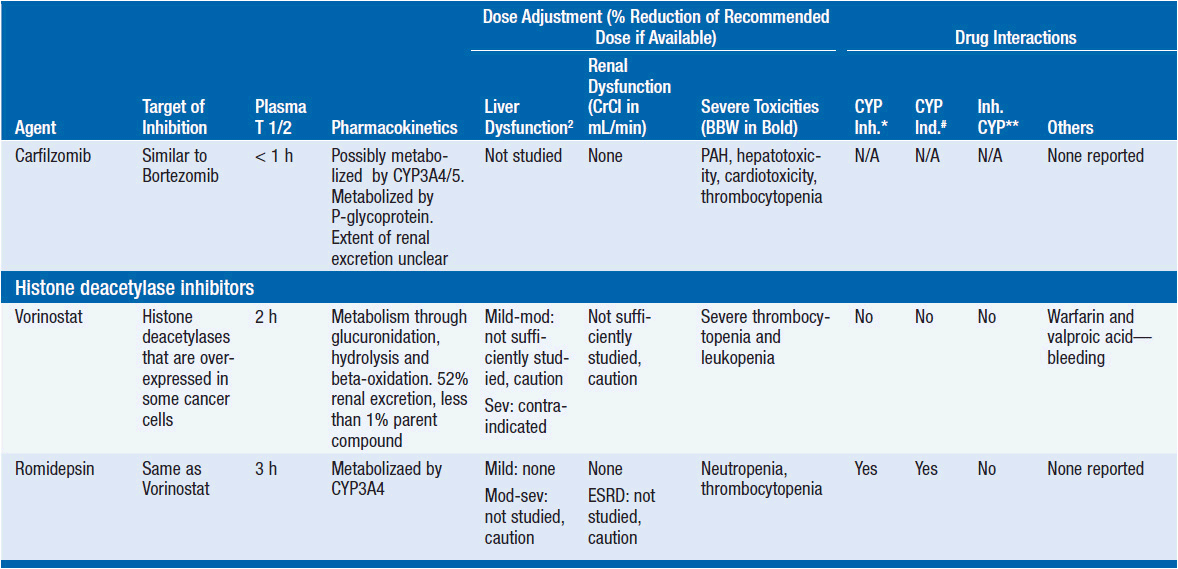
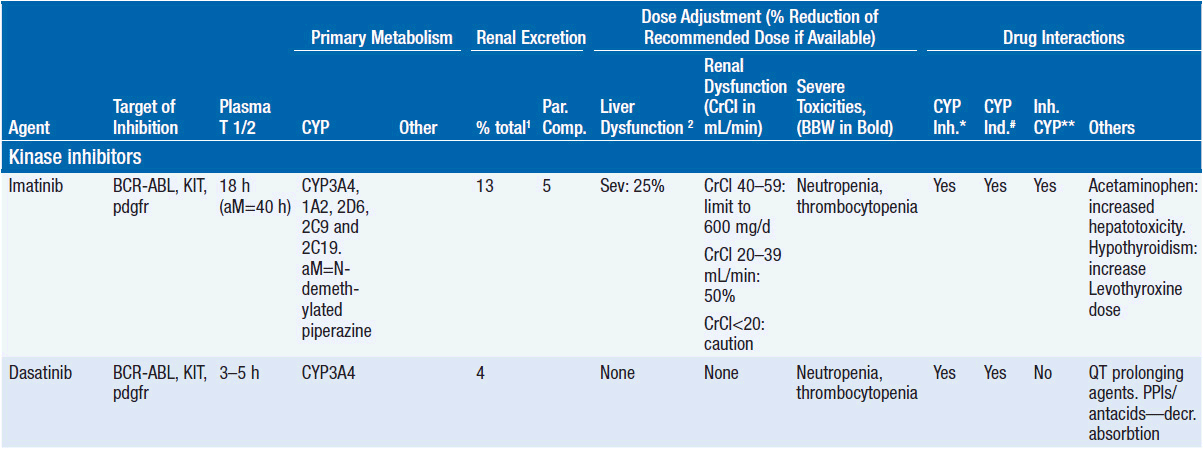
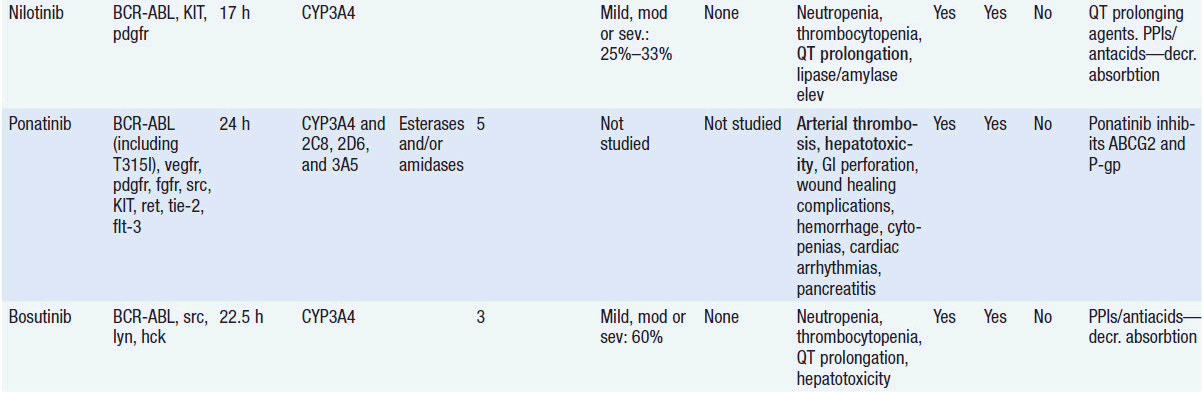
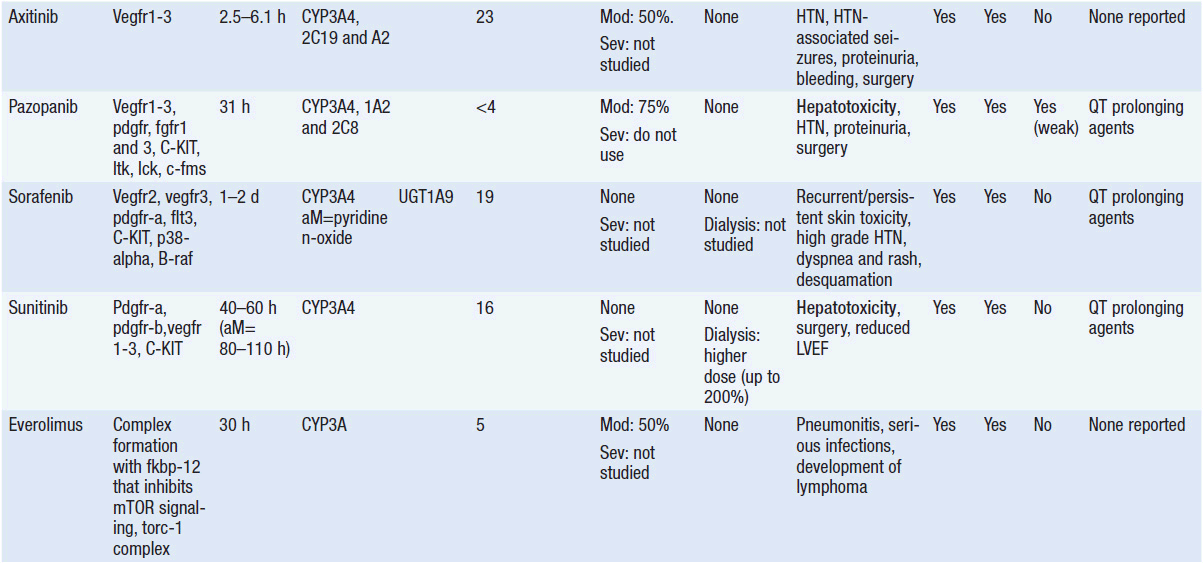
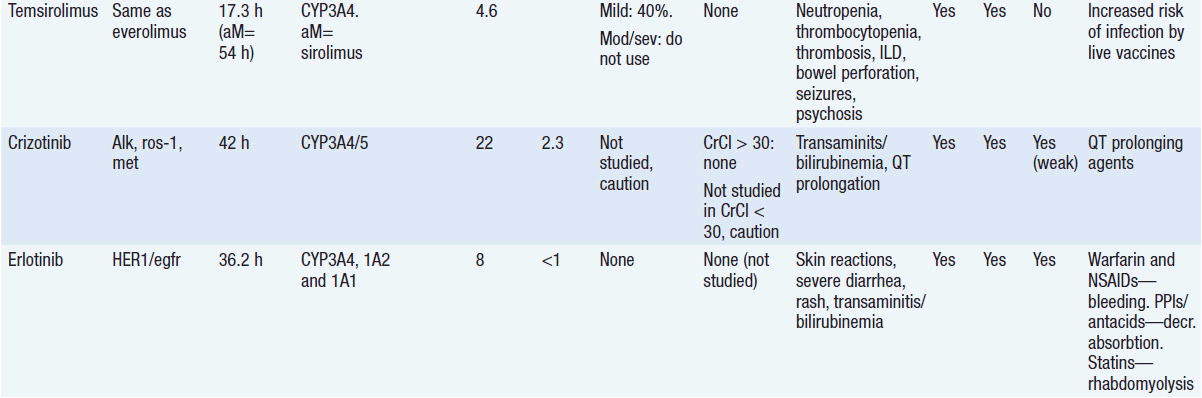
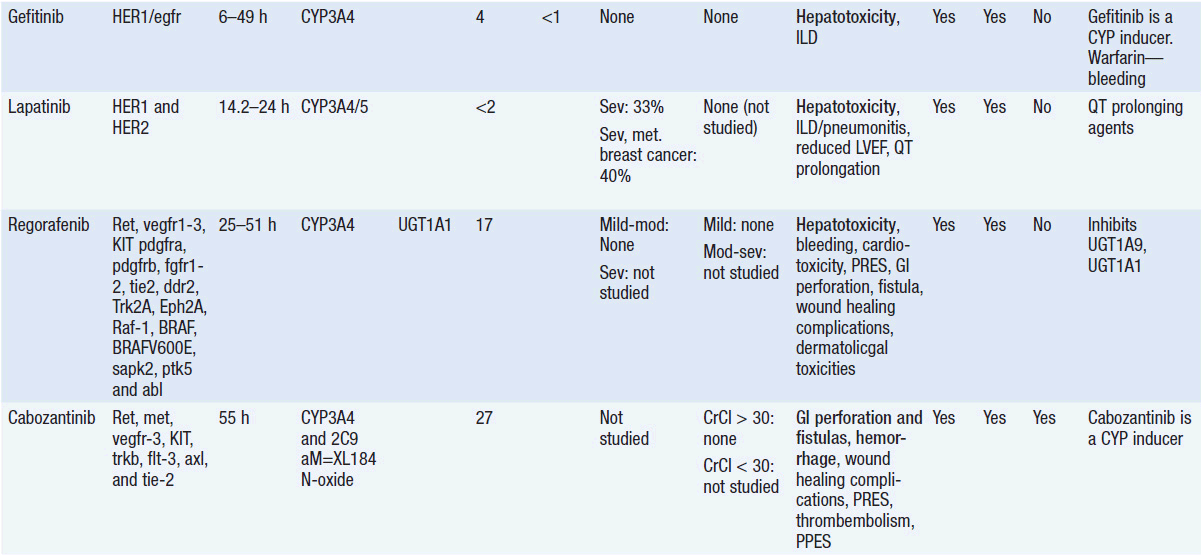
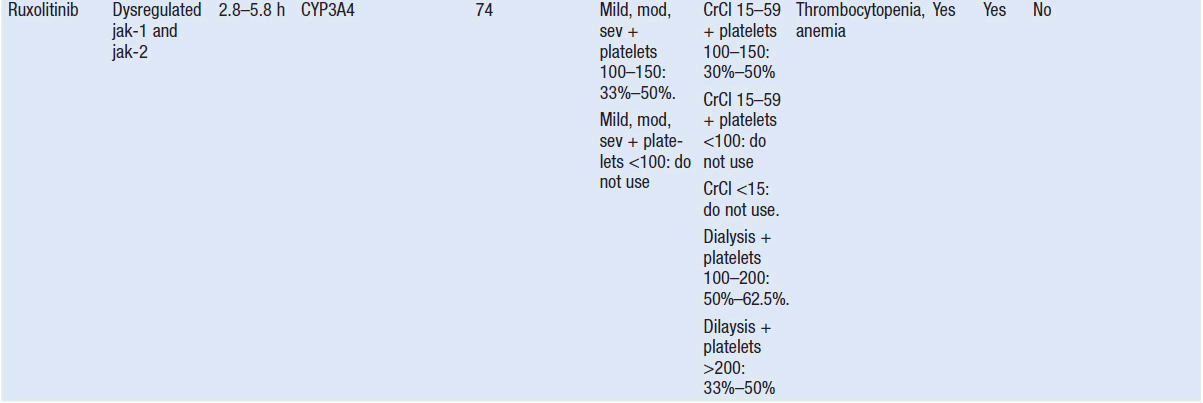
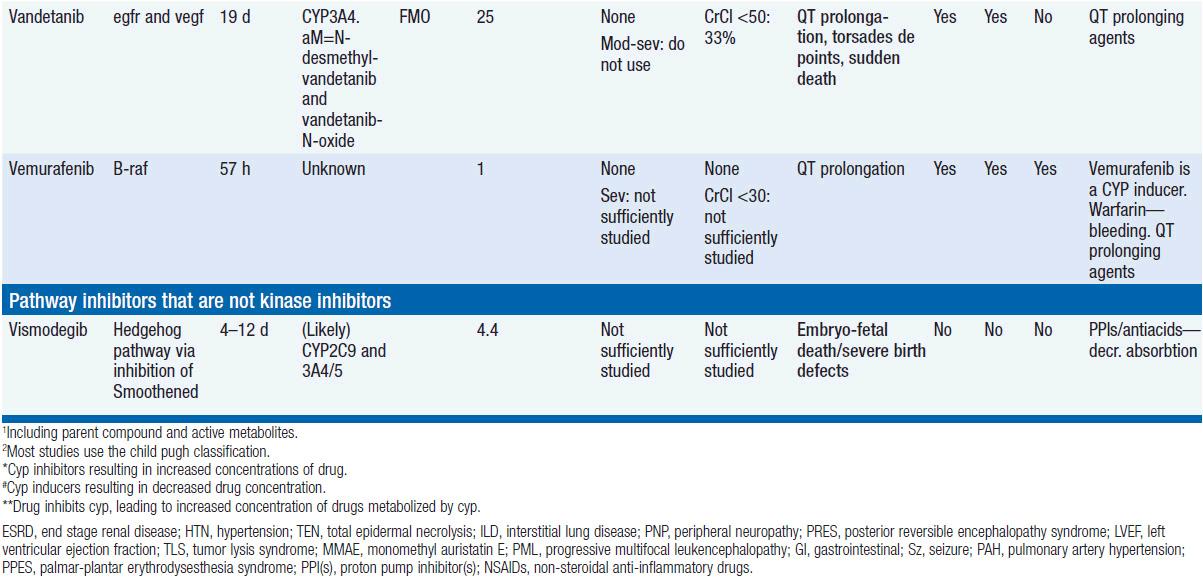
Important discoveries have revealed the molecular basis for the transformation, proliferation, and survival of cancer cells. These advances have revealed new targets for cancer drug design, and have produced agents that inhibit the signaling molecules and pathways responsible for cancer (Figure 10-1) (1). These inhibitors of cancer-associated targets include monoclonal antibodies (mAbs) either alone or coupled with cytotoxic agents or radioisotopes; modified proteins and peptidomimetic molecules; and small-molecular-weight drugs. Still in the development stage are small interfering RNAs (siRNA), antisense oligonucleotides, gene therapy approaches, and ribozymes or DNAzymes. In this chapter we will consider the small molecules that have been approved for clinical use. Monoclonal antibodies and their conjugates will be considered elsewhere (Chapter 15) (see Table 10-1) (2–9).
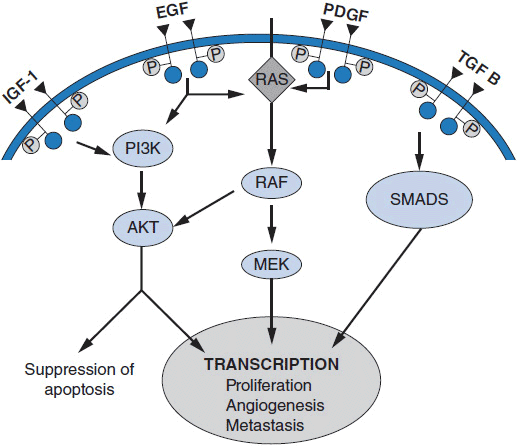






MOLECULAR TARGETS IN CANCER
A rational approach to therapeutic discovery is based on our rapidly growing knowledge of pathways and proteins essential for cancer cell survival, growth, and metastasis. These pathways may be qualitatively unique to cancer (Figure 10-1), or may be simply overexpressed or amplified wild-type proteins. Mutant genes, unique to cancer cells, are particularly attractive in that they alter critical cellular functions and lead to uncontrolled growth, inhibition of apoptosis, escape from growth suppression, invasion of surrounding normal tissues, modifications of the tumor microenvironment including angiogenesis, and metastasis. Inhibition of these mutant functions leads to cancer cell death.
Alterations in a number of fundamental cellular processes may lead to malignant transformation and uncontrolled growth. Malignancy may result from overexpression or amplification of growth factors or their receptors, such as activating mutations or amplifications of the epidermal growth factor receptor (EGFR) family; activation of critical intracellular phosphorylating enzymes such as B-RAF (e.g., by mutation); modulation of the tumor microenvironment such as by activation of angiogenic pathways (e.g., vascular endothelial growth factors [VEGFs] and their receptors); changes in metabolism such as occur with mutations in glucose utilization (such as mutations in the IDH1 or IDH2 genes); epigenetic changes; or activation of anti-apoptotic pathways such as overexpression of Bcl-2 or decreased BAX expression.
 MUTATIONS AND IMPLICATIONS OF “DRIVER GENES” IN CANCER CELLS
MUTATIONS AND IMPLICATIONS OF “DRIVER GENES” IN CANCER CELLS
Certain mutations in cancer cells, particularly those that activate cell receptor tyrosine kinases or downstream proteins involved in signaling within cells, can provide the primary stimulus for cell proliferation and survival. Examples of mutations or translocations in receptors leading to uncontrolled cancer cell proliferation include C-KIT mutations in gastrointestinal stromal tumors (GIST), and EGFR mutations or EML-4/ALK translocations in subsets of non-small cell lung cancer (NSCLC), primarily with adenocarcinoma histology. Mutations of genes in intracellular signal transduction pathways can also become drivers for malignant cell growth or survival. The prototypic example of this is the BCR-ABL translocation in chronic myelogenous leukemia (CML). Another cogent example is the constitutive activation of B-RAF, a protein in the RAS-RAF-MEK pathway, by mutation in melanoma (3, 4). These mutations create “addiction” to the continuous signaling, so that when signaling is blocked, the cancer cells die. In experimental settings, siRNAs directed against these mutant genes turn off the survival signals and lead to cell death, while small molecular inhibitors of the offending kinase cause tumor cell death (3, 4, 8, 9) in human subjects. The first agent to target the RAS-RAF-MEK pathway in human cancer is vemurafenib, which inhibits the V600E mutant form of the B-RAF kinase in melanoma (3, 8, 9). Interestingly, activating mutations of B-RAF are also found in colon, lung, and a number of other cancers, although these mutations have not been as responsive to vemurafenib as melanomas, indicating that the cellular context matters. Inhibitors of PI-3 kinase and its downstream signaling partner mTOR have led to beneficial treatment of renal cancers and neuroendocrine tumors (4). Multiple agents specific for isoenzymes of PI-3 kinase itself are undergoing evaluation in breast cancer, lymphoma, and endometrial cancer.
Mutations in tumor suppressor genes such as p53, retinoblastoma (RB), or the phosphate and tensin homolog (PTEN) that regulates the PI-3 kinase pathway can produce loss of important brakes on proliferation and enhance cell survival, leading to transformation of normal cells as well as contributing to the prolonged survival of cancer cells. These changes have proved harder to target because it is more difficult to return normal function to a protein than to inhibit aberrant function. However, new approaches are being explored aimed at indirectly reversing adverse effects of suppressor gene mutations such as by modulating downstream effectors of the mutant proteins or by targeting epigenetic factors or miRNAs important in control of functions of the tumor suppressor gene (10).
In the following, we discuss several specific examples of clinically effective targeted therapies.
1. Inhibitors of growth factor receptors or their ligands. Growth factor receptors and their ligands are overexpressed or amplified in many epithelial malignancies and are mutated in others (2, 3, 6-8). They are essential for promoting proliferation, survival, and metastasis of various kinds of cancer. The expression of mutated receptors on the cell surface and the presence of their ligands in the circulation make these altered pathways accessible to monoclonal antibodies. Examples of growth factors and receptors currently being effectively targeted include the following:
• EGFR family, including HER1 (EGFR), HER2, and HER3. EGFRs are present on normal epithelium and overexpressed in many cancers and mutated in a subset of NSCLC. The majority of NSCLC with mutated EGFR respond to anti-EGFR therapy, either drugs or antibodies. Anti-EGFR therapy with antibodies has also proved useful in colorectal and head and neck cancers in which the EGFR is not mutated. Amplified and overexpressed HER2 is a major target for a subset of breast and gastric cancers.
• ALK: Activating translocations were originally identified in anaplastic large cell lymphoma. Subsequently the mutated and translocated receptor was shown to respond to crizotinib in a subset of NSCLC adenocarcinomas as well as in patients with inflammatory myofibroblastic sarcoma. Activating mutations of ALK are also present in a subset of patients with neuroblastoma. A closely related receptor, ROS1 kinase, is translocated in a small subset of NSCLC and is susceptible to inhibition by crizotinib.
C-KIT mutations are frequently found in GIST tumors and uncommonly in several other neoplasms, including mucosal melanomas and mast cell disease.
• VEGF/VEGFR: These play an important role in tumor-associated angiogenesis for many epithelial and mesenchymal tumors, and for primary brain tumors.
Various strategies have been employed to inhibit function of these receptor pathways. Responses are well documented in patients treated with mAbs to HER2 (e.g., trastuzumab) or EGFR (e.g., cetuximab), and small-molecular-weight inhibitors (e.g., erlotinib for EGFR mutated NSCLC) (2, 3, 6). Bevacizimab, a monoclonal antibody directed against VEGF, is effective either alone for the treatment of renal cell cancer (RCC) or glioblastomas or combined with chemotherapy for colorectal or lung cancer (7). Small molecular inhibitors (such as axitinib or pazopanib) of VEGFRs have also proved effective in RCC (7) and in treating soft tissue sarcomas, while numerous small-molecular-weight drugs effectively block receptor tyrosine kinases.
2. Inhibitors of signal transduction. A number of signaling pathways downstream of growth factor receptors transmit aberrant growth signals and play essential roles in malignancies. These include the RAS-RAF-MEK pathway, the PI-3 kinase pathway, and the NF-kB pathway. Targeting of mTOR (in the PI-3 kinase pathway) is useful in treating renal cancer, breast cancer, and neuroendocrine tumors of gastrointestinal origin. PI-3 kinase inhibitors, some with broad activity against multiple PI3K isoenzymes while others that are specific for specific isoenzyme, are in active clinical development. Inhibitors of the mutated JAK2 oncoprotein, an important signal mediator in myeloid malignancies, are effective in myeloid metaplasia and other myeloproliferative diseases.
3. Inhibitors of cell cycle control. Many of the currently available cytotoxic agents inhibit DNA synthesis, and cell division, but display limited specificity for cancer. Approaches targeting specific overexpressed or otherwise aberrant cyclin-dependent kinases and other cell cycle regulatory proteins are currently being evaluated based on better understanding of the roles that these play in specific malignant cells.
4. Promoters of apoptosis. Apoptosis, or cell death, is dependent on the balance of activity of pro- and anti-apoptotic proteins. This balance is shifted in favor of anti-apoptotic proteins in many neoplastic cells, as for example the BCL-2 protein activated in follicular lymphoma. Inhibitors of the BH3 family of anti-apoptotic proteins have demonstrated activity against chronic lymphocytic leukemia, and continue in clinical evaluation, alone, combined with other targeted agents, and in combinations with chemotherapy.
5. Restoration of the function of tumor suppressor proteins. Research continues on approaches aimed at restoring normal functions of these critical proteins (p53, p21) to cancer cells (5, 11). Since restoration of the function of the proteins themselves has so far proved intractable, most of the current emphasis is aimed at indirectly modulating this function (see above for further discussion).
7. Inhibition of telomerase. Although much has been learned about the role of telomerase in maintaining telomeres in neoplastic cells and thus allowing continued survival and proliferation, significant preclinical work is needed to translate this into a useful anticancer approach. The furthest along of the approaches directed at telomerase is a vaccine targeting a portion of the telomerase protein. This vaccine is currently being evaluated in a phase III trial in combination with chemotherapy for metastatic pancreatic cancer (12).
8. Inhibitors of chromatin modifiers and epigenetic factors. Methylation of DNA or histones, acetylation of histones, and production of micro-RNAs, and long noncoding RNAs all modify gene expression and differentiation in normal and malignant cells (5, 11). Examples of approved agents targeting epigenetic factors include the histone deacetylase inhibitors (vorinostat, romidepsin), which have activity against cutaneous T-cell lymphoma (CTCL), and inhibitors of DNA methylation (azacytidine) in myelodysplasia.
9. Inhibitors of metabolism. Abnormal dependence on glycolysis has long been known to be an important aspect of malignant cells. The past decade has revealed other dramatic metabolic differences in tumors, including enhanced utilization of glycine or glutamine and activation of a number of enzymes (e.g., IDH-1 and 2, DNA methyltransferase, altered pyruvate kinase). These and other metabolic processes have become targets for drug development (13).
While single specific targets may initiate malignant transformation, whole genome sequencing of tumors has revealed that most cancers contain multiple mutations. In most cases, the role of these additional mutations in drug resistance and survival is unknown. In addition there are important influences on tumor biology coming from the tumor microenvironment. Thus, targeting multiple genes and their protein products may be necessary in order to kill the heterogeneous clones of cells within any given cancer. Recent studies have confirmed the marked heterogeneity of mutations and other changes in different cells within the same malignancy (14). Thus, targeting multiple proteins or pathways, either through multitargeted single inhibitors, such as sunitinib, sorafenib, or regorafenib, or through combination strategies (the combination of B-RAF and MEK inhibitors in melanoma) (15), will likely be required to maximize antitumor efficacy. In addition, targeting the environment is also important, as demonstrated by activity of antiangiogenic agents (5, 7).
CURRENTLY APPROVED TARGETED AGENTS IN CANCER THERAPY (USA)
 SMALL MOLECULES
SMALL MOLECULES
Because they can easily be subjected to high throughput screening and readily modified to incorporate favorable pharmacologic properties, small molecules remain the most attractive and straightforward class of agents for targeted therapy (3, 4, 6-9). High-throughput screening against recombinant proteins allows identification of lead compounds with affinity for the specific target of interest. Subsequent preclinical evaluation, using crystallography and in vitro testing, and analogue chemistry, yields compounds of high target affinity and specificity, with favorable drug properties (e.g., oral bioavailability, extended plasma half-life, decreased toxicity). The major pharmacological properties of representative approved targeted small molecules (grouped by target) are discussed below and are summarized in Table 10-1, which provides information of targets, pharmacokinetics, toxicity, and drug interactions.
TARGET: BCR-ABL
Mechanism of action: The 9:22 translocation in CML places the ABL tyrosine kinase gene on chromosome 9 in juxtaposition to the breakpoint cluster region (BCR) of chromosome 22 with a resultant protein that has constitutive phosphorylating activity, activating multiple downstream signaling pathways and leading to enhanced cell proliferation and survival. Like most other kinase inhibitors, imatinib competes for the ATP binding site of its target protein leading to potent inhibition of the tyrosine kinase activity (8
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


