Figure 14-1 Antagonistic pleiotropy This concept states that signals that engage the cell cycle also engage cell death, and tissues therefore expand only if cell death is blocked. Note that this does not mean that the cell cycle, or any component of its machinery, is responsible for cell death. Survival signals can either be cell intrinsic or extrinsic and need only be specific for the death mechanism engaged by the original growth-promoting signal. The term antagonistic pleiotropy is borrowed from a theory of aging of the same name, which on the surface is very different from the concept of tumor suppression as used here. There are, however, interesting parallels. In the aging theory, genes that are beneficial early in the life of an animal will be selected, even if their activities are detrimental later in life. The reader might wish to consider how this concept may apply to aging and cancer and how these ideas relate to the use of the term here.
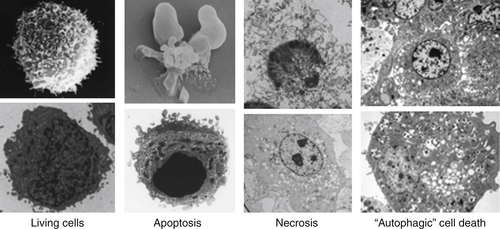
Figure 14-2 Types of cell death Electron micrographs of cell death showing features of each. See text for explanation. NOTE: All figures except as noted are from DRG. Credits: Living and apoptotic cells, DRG and Yufang Shi; necrotic cells, DRG and Nigel Waterhouse; and autophagic cell death, from Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118:79-88.
From the perspective of cancer biology, however, the distinction is particularly useful. When cell death is engaged as a mechanism of tumor suppression, it may be regarded as suicide, whereas the effects of therapeutic intervention may manifest either by recruiting such a suicide pathway or by effectively sabotaging a process in the cancer cell to result in its death.
The best example of active cell suicide is apoptosis, although there may be others (as we note in Figure 14-3 and in the following discussion). In contrast, the lethal overproduction of reactive oxygen species (ROS) by disruption of the mitochondrial electron transport chain, the engagement of membrane NADH oxidases, or the loss of ROS scavenging mechanisms may be regarded as cellular sabotage (see Figure 14-3, and other examples discussed later).
This distinction blurs when we consider the evolution of cell death. Any mechanism of cellular sabotage could well be selected to become a process of cellular suicide, and indeed, many scenarios for the evolution of apoptosis begin with this premise. At this point, it may be best to remind the reader that these distinctions are, like many classifications, merely a starting point for understanding the intricacies of the processes. Nevertheless, it might be interesting to think about why, of all the possible cell suicide mechanisms that might have evolved, apoptosis has emerged as the predominant form of active cell death in the animal kingdom.
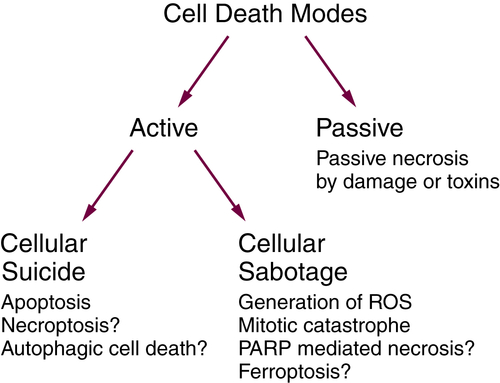
Figure 14-3 Active and passive cell death See text for explanation. PARP, polyADP-ribose polymerase; ROS, reactive oxygen species.
Apoptosis
Caspase Activation
Apoptosis is orchestrated by the actions of caspases, which are cysteine proteases (that is, they have a cysteine at the active sites). There are at least 17 caspases that have been described in mammals, and we understand the functions of only a few of them. The process of apoptosis occurs through the activation of a subset of these, called the executioner caspases (caspase-3, caspase-6, and caspase-7 in mammals), and once activated, these cut up to a thousand different cellular substrates in the cell to precipitate the changes associated with this form of cell death. The “cuts” are at specific sites in the substrates and occur at aspartate residues. This is the origin of “caspase,” a cysteine protease with Asp-ase activity.
The executioner caspases are present in cells in an inactive, dimeric form, which becomes activated when cut at specific aspartate residues, resulting in a conformational change that forms the active enzyme. The active enzyme now cuts its substrates, some of which are responsible for the changes associated with apoptosis. 6
In general, this activating cleavage is mediated by other caspases, called initiator caspases. Unlike the executioner caspases, the inactive forms of the initiator caspases present in cells are monomeric and contain large prodomains that include sites for protein-protein interaction. Adapter molecules, which form “caspase activation platforms,” bind to the prodomains of initiator caspases, forcing them into contact, and the latter then fold into active enzymes. These can then process the executioner caspases, thereby activating them to cleave their substrates and precipitate apoptosis.
The formation of caspase activation platforms and the caspases they activate define the apoptotic pathways. Here we consider in detail only two major pathways of apoptosis, the mitochondrial pathway and the death receptor pathway. It should be noted, however, that there are others, reviewed elsewhere. 7 One of these involves the formation of caspase activation platforms for the engagement and activation of caspase-1, which is critically involved in inflammatory responses by processing specific cytokines and promoting their release. The activation of caspase-1 can also kill cells by a process resembling apoptosis (often called pyroptosis). The activation of caspase-1 occurs in response to many infectious agents and has been implicated in promoting forms of cancer that are associated with inflammatory conditions. Caspases can directly signal apoptosis or can use mitochondria as an intermediate, and additional point of regulation, in signaling of apoptosis.
The Mitochondrial Pathway of Apoptosis
Starting in the 1980s, the intracellular molecules that controlled the morphology referred to as apoptosis began to be identified. 8,9 Cell death had previously been thought of as a passive event (as noted earlier): a universally negative occurrence that cells were constructed to avoid at all costs. Careful observations in human pathological samples as well as in model systems revealed that instead, cell death in multicellular organisms was often a carefully controlled event with stereotypical morphologic and temporal patterns. Moreover, when it was observed that altering the function of certain genes could alter the commitment, phenotype, and progression to cell death, it became clear that cells contained within them genetic programs to perform a function of choosing death as a cell fate, and then committing cell suicide. Although several different forms of programmed cell death have been identified, the first to be characterized was apoptosis.
The mitochondrial (sometimes called intrinsic) pathway of apoptosis responds to a diverse group of initiating events, including treatment with a wide variety of cytotoxic drugs, growth factor withdrawal, and oncogene activation. The mitochondrial pathway of apoptosis get its name because of the centrality of this organelle in coordinating both cell fate decision making as well as execution. Key features of apoptosis via the mitochondrial pathway include mitochondrial outer membrane permeabilization (MOMP), followed by release to the cytosol of molecules that facilitate execution of the death program, including cytochrome c, SMAC, and Omi. Cytochrome c is essential to the formation of one of the caspase activation platforms, the apoptosome, which is formed when cytochrome c activates APAF-1 to oligomerize, and the latter then binds and activates caspase-9. 10 Although caspases may perform normal physiological functions, they are best known for cleavage of proteins during apoptosis important for cell integrity, including cytoskeletal proteins and PARP-1. Among the caspase-dependent phenomena commonly observed in association with apoptosis are extracellular exposure of the phosphatidylserine that is usually found only on the inner leaflet of the plasma membrane (which results in recognition by Annexin V) and internucleosomal cleavage of chromosomal DNA (which results in laddering of DNA on electrophoresis and nuclear condensation). In vivo in a multicellular organism, an important consequence of these changes is recognition and clearance of the apoptotic cell by phagocytic cells. It may well be that commitment to cell death is already made at the point of MOMP, whereas many of the downstream morphologies we associate with apoptosis might be most important mainly in facilitating the clearance of apoptotic cells, minimizing inflammation, and optimizing cannibalization of their macromolecules. 11–13
The BCL-2 Protein Family
The B cell leukemia/lymphoma 2 (BCL-2) family of proteins controls commitment to apoptotic cell death via the mitochondrial pathway. 9 The discovery and characterization of this important family is intimately linked to cancer, as BCL-2 was initially cloned from the t(14;18) chromosomal translocation that is present in nearly all cases of follicular lymphoma. 14–16 The translocation places the bcl-2 gene on chromosome 18 under the control of regulatory elements of immunoglobulin genes, yielding overexpression of the BCL-2 protein in cells of the B-lineage.
The key step in apoptosis commitment that is controlled by the BCL-2 family is MOMP. 17–19 In simplest terms, there are pro- and anti-apoptotic members of the BCL-2 family. When the pro-apoptotic proteins overwhelm the anti-apoptotic ones, MOMP occurs, and the cell is irreversibly committed to apoptosis. The details of this commitment involve complicated interactions among the BCL-2 family proteins that number greater than a dozen. BAX and BAK are obligate pro-apoptotic “effectors,” proteins that homo-oligomerize to form the pores that cause MOMP. 20 In order to form the oligomers required to cause MOMP, BAX and BAK must be activated. 21,22 Proteins of the pro-apoptotic “activator” BH3-only protein subfamily, which include BIM, BID, and perhaps PUMA, can directly interact with BAX and BAK to cause an allosteric change that functionally corresponds to activation. There may also be other ways that BAX and BAK are activated, perhaps even independent of interaction with other proteins. 23
Anti-apoptotic BCL-2 family proteins include BCL-2, BCL-XL, BCL-w, MCL-1, and BFL-1 (A1). These proteins inhibit apoptosis by binding and sequestering activator proteins or monomeric “activator” BAX or BAK proteins before they can oligomerize. 24,25 Another subfamily, the pro-apoptotic “sensitizer” BH3-only proteins, lack the ability to directly activate BAX and BAK with high efficiency, but promote apoptosis by competitively inhibiting the ability of anti-apoptotic proteins to bind activators, BAX, or BAK (Figure 14-4 ). 21
BH3-only proteins get their name from their possession of the BCL-2 homology 3 (BH3) region, but of no other BCL-2 homology region. 26 The BH3 domain is essential for the pro-death activity of all of the pro-apoptotic BCL-2 family proteins. It is a roughly 20 amino acid amphipathic α-helix that binds into a hydrophobic BH3 binding cleft present in all of the anti-apoptotic proteins. There is a selective pattern of interaction of BH3-only proteins with anti-apoptotic proteins. 24,27–29 For instance, the BAD BH3 domain interacts selectively with BCL-2, BCL-XL, and BCL-w, whereas the NOXA BH3 peptide interacts selectively with MCL-1. Small-molecule BH3 domain mimetics that compete for these interactions, effectively inhibiting anti-apoptotic protein function, are being tested in cancer clinical trials (see later discussion). 30
Mitochondrial Priming and Chemotherapy Response
The concept has emerged from many studies, especially in murine models of cancer, that apoptosis serves as a natural barrier to carcinogenesis. 31,32 In several murine models, defects in apoptosis facilitate carcinogenesis, supporting this concept. 33–35 Many of the changes that commonly occur in oncogenesis, including genomic instability and oncogene activation, contribute to pro-apoptotic signaling, which may well result in deletion of nascent malignant clones. Malignant clones that do survive must therefore have selected for mechanisms of evading apoptotic death provoked by this pro-apoptotic signaling by some combination of fostering anti-apoptotic signaling and attenuating pro-apoptotic signaling.
However, as it is commonly used, the oft-made assertion that resistance to apoptosis is a fundamental property of all cancer begs the question, “Compared to what?” It is difficult to find experimental or clinical evidence that cancers generally are more resistant to apoptosis than the normal nonmalignant cells in the human body. The fact that cancer cells have successfully buffered prior pro-apoptotic signaling does not necessarily mean that they are well prepared to evade subsequent pro-apoptotic signaling.
In fact, many conventional chemotherapies used in the clinic kill cells via apoptosis. The specific mechanisms may vary widely. Taxanes may increase levels of BIM, alter subcellular localization of BH3-only proteins, and decrease levels of MCL-1. 36–38 Cytotoxic agents can induce a decrease in MCL-1 levels, though whether this is a necessary and sufficient step for commitment to apoptosis in these cases is less clear. 39 DNA-damaging agents, by activating p53, can activate transcription of pro-apoptotic PUMA, NOXA, and BAX (see also Chapters 3 and 15). 40–43 In addition, p53 may promote apoptosis via direct interaction with BCL-2 family proteins. 44,45 Space does not permit detailing all of the relationships that have been discovered between conventional chemotherapy agents and the BCL-2 family. Although there are sometimes attempts to identify the key BCL-2 family dictating response to a particular agent, it is likely that following most chemotherapies, more than one BCL-2 family protein takes part in determining the cell fate decision.
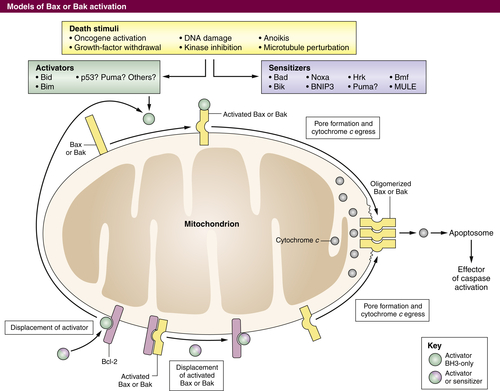
Figure 14-4 Control of MOMP by the BCL-2 family of proteins MOMP occurs following homo-oligomerization of activated BAX and/or BAK into a pore (right), permitting release of proteins such as cytochrome c that facilitate caspase activation. BAX or BAK pore formation can be caused by the displacement of monomeric BAX or BAK that is already activated from anti-apoptotic proteins. This displacement can be effected by BH3-only proteins or BH3 mimetics (bottom). Alternatively, in response to a variety of stimuli, an increase in activator proteins such as BIM or BID can activate BAX or BAK, which can then homo-oligomerize (top). Sensitizer BH3 proteins or mimetics can also displace activators from anti-apoptotic proteins to effect BAX or BAK activation (left).
The ability of chemotherapy to selectively kill cancer cells makes it likely that in many cancer cells the apoptotic pathway is not only intact, but even more sensitive to pro-apoptotic signaling than in normal cells. This hypothesis has been directly tested using a mitochondrial assay called BH3 profiling, an assay that systematically compares mitochondrial response to a standardized panel of synthetic BH3 domain peptides. 46,47 These studies have shown that chemosensitive cancer cells are indeed more sensitive to pro-apoptotic signals than normal cells, suggesting mitochondrial apoptotic priming as an important determinant of the therapeutic index in vivo and in vitro. Normal hematopoietic cells are the most primed of normal tissues, consistent with their perennial role as the site of dose-limiting toxicity for most cytotoxic regimens. Measuring pretreatment mitochondrial priming may even be useful as a predictive biomarker, because it correlates well with clinical response. 46,47
From a perspective of protein biochemistry, cells that are highly primed have little anti-apoptotic reserve to buffer subsequent pro-apoptotic signaling. Very often, a highly primed cell will express abundant anti-apoptotic BCL-2 family proteins, but these will already be occupied by pro-apoptotic activators such as BIM. 24,48,49 In these cases, the cells are dependent on the continuous function of the anti-apoptotic protein(s) for survival. Inhibition of one or more of these can result in MOMP and apoptosis in highly primed cells (Figure 14-5 ).
It is worth considering the distinction between expression of an anti-apoptotic protein, and dependence on that anti-apoptotic protein. If one enforces BCL-2 overexpression via transfection of a plasmid vector in a healthy cell line already well established in culture, one will not necessarily obtain a BCL-2–dependent cell line, because there is likely no dependence on the additional BCL-2. The cell line was doing fine without it before transfection, and inhibition of BCL-2 would just restore the status quo ante. In oncogenesis, however, increased expression of BCL-2 is selected for, not extrinsically induced. This implies that the increased BCL-2 that was selected for is required for the continuous buffering of pro-apoptotic signals. In biochemical terms, that means that BCL-2 is largely already in complex with pro-apoptotic BCL-2 family members such as BIM or BAX. In the instance of the transfected cell line, however, BCL-2 is mainly empty and ready to buffer subsequent pro-apoptotic signaling. In the case where BCL-2 expression has been positively selected in the face of extant pro-apoptotic signaling, the BCL-2 is mainly full. Rather than providing anti-apoptotic reserve, the BCL-2 in the latter case is actually storing pro-apoptotic proteins at the mitochondrion, priming it for sensitivity to apoptosis. 24 The cancer cell just discussed is thus dependent on continuous BCL-2 function for survival. In other cancer cells, similar to the transfected cell line, BCL-2 expression may be incidental, rather than selected for, and the BCL-2 not required tonically. Protein and transcript levels of multiple BCL-2 family members, as well as functional assays such as BH3 profiling, have been used to identify cells that are dependent on BCL-2 and hence likely to be sensitive to antagonists of BCL-2 (see later discussion). 48,50,51
Targeted Therapies and Apoptosis
In the past decade, modern “targeted” therapies have occupied the lion’s share of attention in preclinical development, with several achieving approval for clinical use in cancer. Most of these agents also kill by inducing signaling via the mitochondrial apoptotic pathway. However, in this case, selective killing of cancer cells depends less on differences in pretreatment mitochondrial priming and more on selective dependencies of cancer cells. For instance, imatinib, the first small-molecule kinase inhibitor to be approved for use in cancer, inhibits activity of the Abl kinase, an activity that is enhanced by the BCR/ABL fusion protein created by the t(9;22) chromosomal translocation present in chronic myelogenous leukemia (CML). The selective killing of malignant CML cells over normal cells depends on the selective dependence of CML cells on activity of the BCR/ABL kinase. 52 Another family of tyrosine kinase inhibitors, those directed against epidermal growth factor receptor (EGFR), are selectively effective in lung cancers with activating mutations in EGFR. 53–55
A general property of kinase inhibitors in cancer, whether tyrosine kinase or serine/threonine kinase inhibitors, is their utilization of the mitochondrial apoptotic pathway to kill cancer cells. Interestingly, although doubtless other BCL-2 family members also contribute, BIM, a pro-apoptotic activator BH3-only protein, is regulated by many different kinase inhibitors. In turn, lethality of kinase inhibition depends on the upregulation of BIM. BIM upregulation is required for killing by imatinib whether in CML or in gastrointestinal stromal tumors (GIST) where the constitutively active c-KIT kinase is the relevant target. 56,57 Similar observations have been made for inhibitors of EGFR, MEK, ALK, and b-RAF. 37,58–61 A fascinating human genetic study found that a germline polymorphism of BIM that removes the BH3 domain that is required for pro-apoptotic function is common in an Asian population. 62 Intriguingly, this polymorphism conferred inferior clinical response to inhibitors of both BCR-ABL in CML and of EGFR in non–small-cell lung cancer, apparently confirming its importance in cancer cell–fate decision making in clinical use of kinase inhibitors.
Proteasome inhibitors such as bortezomib have become a mainstay of multiple myeloma therapy and have found application in other diseases such as mantle-cell lymphoma. These also apparently kill via the mitochondrial apoptotic pathway. In this case, the most often cited mechanism of response is the pro-apoptotic BH3-only family protein NOXA, which can selectively bind and inactivate MCL-1, an anti-apoptotic protein also in the BCL-2 family. 63 Of course, proteasome inhibitors alter the levels of thousands of proteins, so that there may be several proteins that are affected that are important for cell-fate decision after proteasome inhibitors.
Directly Targeting the BCL-2 Family in Cancer Treatment
Given the centrality of the BCL-2 family of proteins in determining cell fate in cancer, considerable effort has been expended on inhibition of anti-apoptotic BCL-2 family proteins to induce cancer cell death. 30
These efforts are in their infancy, and important questions need to be resolved. For example, should the agent have a narrow or broad spectrum of inhibition of anti-apoptotic BCL-2 family proteins? Broad-spectrum agents will likely be more toxic to more kinds of cancers; however, there is the possibility of a narrowed therapeutic index that could limit clinical utility. Will these agents be best used as single agents or in combination with others? Although some diseases may demonstrate clinical sensitivity to single agents, it may well be that combination with the powerful, albeit less selective, pro-death signaling induced by conventional cytotoxic agents will be necessary to provide the significant benefit in long-term clinical outcomes that is so badly needed in many cancers. In addition, combinations with targeted agents such as kinase inhibitors have proved promising in vitro.
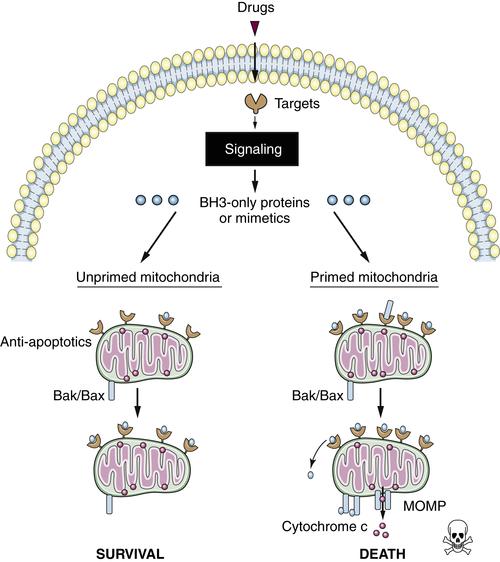
Figure 14-5 Primed versus unprimed mitochondria Functionally, primed mitochondria can be defined as those that are most sensitive to BH3 peptides. Unprimed mitochondria (left) have a relative excess of “empty” anti-apoptotic proteins, affording anti-apoptotic reserve that can buffer subsequent death signaling. Primed mitochondria (right) may have abundant anti-apoptotic proteins, but these are relatively “full,” occupied by pro-apoptotic proteins. Primed mitochondria have little anti-apoptotic reserve and are relatively sensitive to subsequent death signaling.
The Death Receptor Pathway of Apoptosis
The death receptors are a subset of cell surface receptors that are members of the tumor necrosis factor (TNF) receptor (TNFR) superfamily. These receptors can be activated by ligands, which are themselves a subset of the TNF superfamily. The death receptors include one of the receptors for TNF, TNFR1, the TRAIL receptors, and CD95 (also called Fas or APO1).
When a death receptor is engaged by its ligand, this can result in apoptosis. In this death receptor pathway of apoptosis, the initiator caspase is caspase-8, and its activation platform involves the adapter protein, FADD, which binds to the prodomain of the caspase, allowing it to form its active dimer.
The simplest form of this process is seen in the ligation of CD95 (this may apply to the TRAIL receptors as well). Ligation of CD95 causes the exposure of a protein interaction region in the intracellular part of the receptor, which rapidly binds to FADD. Several receptors and the associated FADD form the caspase activation platform, and caspase-8 is recruited to FADD for its activation. The proteolytic activity of the bound caspase-8 now cuts the caspase, removing the prodomain and releasing the now-stabilized dimer (Figure 14-6 , A).
In the case of TNFR1, the process is much more complex. Binding of ligand to TNFR1 induces exposure of the intracellular region of the receptor, but this does not bind FADD. Instead, another adapter, called TRADD, is bound, as well as a number of other signaling proteins, some of which modify the associated proteins with ubiquitin. Among these is RIPK1, a protein kinase, discussed further later. Also among these are proteins involved in the activation of a transcription factor, nuclear factor-κB (NFκB). The initial complex (complex I) is then released from the receptor, and its subsequent signaling outcome depends on additional protein interactions. Among these is a protein called c-FLIPL (also called CFLAR).
c-FLIPL closely resembles caspase-8 but lacks a catalytic cysteine. When complex I is released, TRADD binds to FADD in the cytosol, and this recruits caspase-8. However, if c-FLIPL is present, the complex that forms (complex IIa) contains heterodimers of caspase-8 and c-FLIPL, and this does not promote apoptosis (discussed later). However, if the activity of NFκB is blocked or disrupted, or if the expression of c-FLIPL is otherwise blocked, complex IIa activates caspase-8 (see Figure 14-6, B). Another complex that forms in cells exposed to TNF involves the kinase RIPK1 (complex IIb). Like TRADD, RIPK1 can bind to FADD and form a caspase-activation platform. Again, if c-FLIPL is present, caspase-8–c-FLIPL heterodimers form, and apoptosis does not proceed. However, if c-FLIPL is absent, this can also result in caspase-8 activation (see Figure 14-6, C).
The formation of the RIPK1-FADD complex is not restricted to the death receptor pathway. It can form as a consequence of DNA damage, although the precise signaling mechanisms are not fully elucidated. 64 Another way in which this forms is as a result of ligation of some Toll-like receptors (TLRs), molecules that recognize components of infectious organisms, such as bacteria. Some TLRs recruit a signaling molecule, called TRIF, and this can engage RIPK1 to form a complex with FADD. 65 As with the other cases, the subsequent activation of caspase-8 to promote apoptosis depends on the presence of c-FLIPL. Efforts to therapeutically activate death receptor pathways for cancer therapy are in development. 66
Mitochondrial Pathway Activation by Caspase-8
When we discussed caspase activation, we noted that initiator caspases, such as caspase-8, can cleave and thereby activate executioner caspases to promote apoptosis. However, in most cell types, the activated executioner caspases are bound by an inhibitor, X-linked inhibitor of apoptosis protein (XIAP), which then ubiquitinates the caspase and promotes its degradation. This prevents apoptosis from proceeding. However, caspase-8 has another substrate in the cell that indirectly overcomes this inhibition. This protein is BID, one of the BH3-only proteins discussed in the last section. When caspase-8 cleaves BID, this BH3-only protein translocates to the mitochondria to promote MOMP.
Although MOMP and the release of cytochrome c can then lead to activation of caspase-9, as we have noted previously, this event is not necessary in this case. Instead, the release of other proteins, such as Smac and Omi, is required, as these bind to and neutralize XIAP. As a result, the executioner caspases cleaved by caspase-8 can now precipitate apoptosis in the cell. This is illustrated in Figure 14-6, D. This explains the otherwise apparently paradoxical observation that anti-apoptotic Bcl-2 proteins can often inhibit apoptosis engaged by the death receptor pathway.
Necrosis
Passive and Active Necrosis
Necrosis (type III cell death) is distinct from apoptosis and autophagic cell death in terms of both morphology and mechanisms. Cells that die by necrosis generally have diffuse nuclei and a loss of organellar structures, as entry of water into the cell causes swelling and often rupture of membrane compartments.
Understanding necrosis is important in cancer biology for several reasons. Most importantly, cancers that contain high levels of necrosis often have a poor prognosis. This may be because such cancers grow so quickly that they outstrip their blood supply. Another important aspect of necrosis is that it is inflammatory: That is, the contents of the dying cells release mediators (DAMPs, see earlier discussion) that trigger inflammatory responses. Inflammation itself damages tissues and can cause cycles of repair and proliferation involving growth factors that can promote oncogenesis and tumor expansion. In many adult cancers, inflammation may be a major component of the process that promotes tumorigenesis.
As we noted earlier, it is useful to distinguish between cell death that is passive versus active, and this applies to necrosis. Passive necrosis occurs when cells are irreparably damaged by external forces, or when processes that are essential for sustaining cellular homeostasis are blocked or disrupted. This can occur as a consequence of mechanical stress or toxic chemicals or as a result of specialized pore-forming peptides or proteins (e.g., some venoms and bacterial toxins, complement, perforin). Because such processes are intuitively fairly obvious, our focus here is on forms of necrosis that are active, that is, depend on cellular processes and events that, if inhibited, preserve the survival of the cell. For the most part, these all correspond to forms of cellular “sabotage,” although as we will see, there is one form of necrosis that might be “suicide.”
Necrosis as an Adjunct to Other Cell Death Modalities
Before embarking on a survey of the mechanisms of active necrosis, we must note that there are processes that can confuse the distinctions between necrosis and other forms of cell death. These are secondary necrosis and caspase-independent cell death, both related to apoptosis, and many forms of autophagic cell death.
As we mentioned, when a cell dies by apoptosis, it is rapidly engulfed by other cells and removed from the system before any loss of plasma membrane integrity. However, if such engulfment does not occur, the dying cells will take on some features of necrosis as plasma membrane integrity is lost. This is called secondary necrosis and can often be distinguished from other forms of necrosis by some features of apoptosis, such as chromatin condensation. Whether or not secondary necrosis is inflammatory, and thus may contribute to some disease states, remains controversial, but there is accumulating evidence that defects in the engulfment and clearance of apoptotic cells can cause pathology arising from the effects of secondary necrosis.
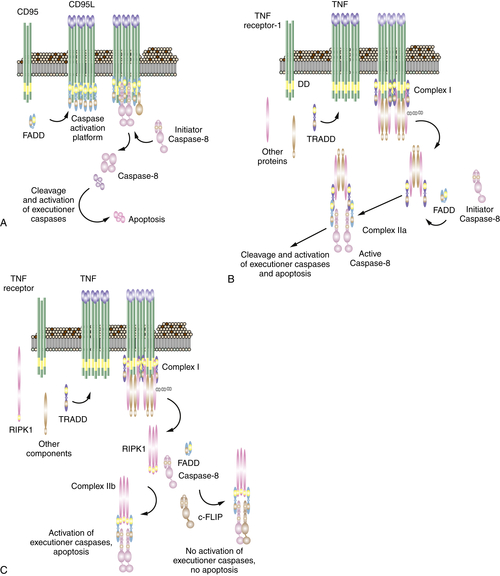
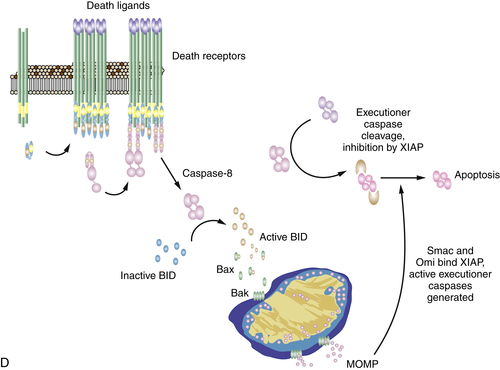
Figure 14-6 Signaling of cell death by death receptors Induction of cell death by (A) CD95; (B) TNF through complex IIa; (C) TNF through complex IIb; and (D) death receptor signaling through mitochondria. The often-used “Fas” to designate CD95 stands for “FS-7 associated surface” antigen, recognized by an antibody capable of inducing apoptosis, originally discovered by S. Yonehara. Because “FAS” designates fatty acid synthase, an unrelated protein, the term has been replaced by CD95. Unfortunately, however, the old nomenclature persists and may cause confusion.
A second form of necrosis that is related to apoptosis is caspase-independent cell death (CICD). 67 This occurs when a cell undergoes MOMP in the mitochondrial pathway of apoptosis, but caspase activation is blocked or disrupted. Although several mechanisms have been suggested for why CICD occurs, an emerging consensus suggests that this is mainly due to energetic consequences of the “mitochondrial catastrophe” produced by MOMP. 68 Notably, regulatory events that control MOMP, such as expression of anti-apoptotic BCL-2 proteins, effectively block CICD and thus distinguish this form of necrosis. Although cells that undergo MOMP but do not engage caspases often die by CICD, cells can survive this event, 69,70 and it is possible that such survival contributes to cancer and/or chemotherapeutic resistance in some cases.
As mentioned earlier, passive necrosis occurs when essential cellular processes are disrupted, but repair mechanisms can insulate a cell from such insults. One major repair mechanism is autophagy (discussed in much more detail later), and therefore a cell that dies as a consequence of such damage may show extensive evidence of autophagy. As a result, we may categorize cells dying in this way as so-called autophagic (type II) cell death. In such cases, however, inhibition or disruption of autophagy will render the cell more susceptible to necrosis, and therefore it may be more appropriate to regard this process as one of necrosis.
Ischemia and Necrosis
Probably the most common cause of necrosis in cancer is as a result of ischemic injury, the loss of blood supply to the tumor. There are two major ways ischemia causes necrosis; the first is via the deprivation of oxygen and nutrients. This occurs in tumors as regions of the mass are deprived of blood supply and can be regarded as passive necrosis. A second, more complex way occurs when blood supply is restricted and then regained, a condition referred to as ischemia/reperfusion injury. Paradoxically, reperfusion of a deprived tissue often results in massive necrosis, and this is a major clinical problem.
Ischemia/reperfusion injury is a complex process we can regard as a form of cell sabotage and thus active cell death, although it is not fully understood. There are three components of ischemia/reperfusion injury that are generally agreed on: a rise in intracellular calcium, the production of reactive oxygen species, and disruption of the mitochondria.
On reperfusion, potassium channels in the plasma membrane open, releasing potassium from the cytosol, and this in turn causes the opening of plasma membrane calcium channels and a rise in intracellular calcium (Ca2+). The increase in Ca2+ has several effects. First, it can activate the protease calpain, which can inflict proteolytic damage to the cell. Pharmacologic inhibitors of calpain have some protective effects in ischemia/reperfusion injury. Second, Ca2+ activates NADPH-oxidases, causing the production of ROS, which can also damage the cell by targeting lipids and DNA. Damage to the DNA elicits activation of polyADP-ribose polymerase (PARP), and extensive activation of PARP can deplete stores of NADH, accelerating death. Scavenging ROS or inhibition of PARP can also produce some protection against this form of injury.
Both Ca2+ and ROS may precipitate necrosis in large part by affecting the mitochondria through a process called the mitochondrial permeability transition. The inner mitochondrial membrane is critical for mitochondrial function, as the control of the distribution of protons and other ions across this membrane drives ATP production and other activities of the organelle. High levels of Ca2+ and/or ROS cause the opening of a “pore” in the membrane that dissipates these critical gradients, and the matrix swells, ultimately destroying the organelle. The components of the permeability transition pore are largely unknown, with one exception: a protein of the mitochondrial matrix, cyclophilin D, has a major role in the process. Animals lacking cyclophilin D display no developmental abnormalities, but are resistant to ischemia/reperfusion injury. Whatever the function of the mitochondrial permeability transition might be, it may be safe to conclude that necrosis induced by ischemia/reperfusion that engages this mechanism represents a form of cellular sabotage. Whether this process can be used to promote cancer cell death therapeutically is not known.
Necroptosis Is a Form of Active Necrosis
In the discussion of the death receptor pathway of apoptosis, a kinase, RIPK1, was introduced, which participates in one of the TNFR complexes and can also function in complexes induced by other means (DNA damage, TLR-TRIF signaling). RIPK1 can also promote a form of necrosis, called necroptosis (to distinguish it from other forms, such as those discussed earlier).
Necroptosis involves the activation of another kinase, RIPK3, which binds to RIPK1 but can also be engaged by other proteins (a putative sensor of viral DNA, called DAI-1, activates RIPK3 and causes necroptosis independently of RIPK1). The activation of RIPK3 rapidly induces cell death with the features of necrosis.
How cell death occurs on activation of RIPK3 is largely unknown and is an area of intense research. One other player in the process is another protein, MLKL, which may itself be a kinase or may act as an adapter to bring specific substrates to RIPK3. Although a number of effector mechanisms have been proposed, including mitochondrial ROS and other mediators, at this point we simply do not know how this form of necrosis occurs.
As discussed in the consideration of death receptor signaling, RIPK1 can recruit the adapter, FADD, into a caspase activation platform that engages caspase-8 and the caspase-like molecule, c-FLIPL. Although the caspase-8–c-FLIPL heterodimer is an enzymatically active protease, it does not promote apoptosis (for reasons that we still do not understand). However, it has now become clear that this activity serves an important function in cells: It blocks the activation of RIPK3 (Figure 14-7 ).
Ligation of TNFR1, or other ways in which RIPK1 is engaged and activated, generally does not cause necroptosis, unless the formation of the FADD–caspase-8–c-FLIPL complex is blocked or disrupted. 71,72 Mice in which the genes for FADD or caspase-8 are deleted die early in embryogenesis, but development is fully rescued by ablation of RIPK3. The effect of ablating c-FLIP is the same, but its rescue is more complex: In this case death occurs only if both RIPK3 and FADD are deleted, most likely because activity of FADD to activate caspase-8 promotes death in cells lacking c-FLIP.
How, exactly, the caspase-8–c-FLIPL heterodimer blocks necroptosis is not fully known. One simple possibility is that it cuts RIPK1 (and perhaps RIPK3), rendering it inactive. However, other possibilities exist, either upstream of RIPK1 or downstream of RIPK3. For example, an enzyme, CYLD, that removes ubiquitin from RIPK1 (required for its necroptotic function) is targeted by caspase-8 73 and therefore may also play a role in the protective function of caspase-8–c-FLIPL.
Why is the system “built” this way? One possibility is that many viruses have evolved mechanisms to inhibit the activation of caspase-8 as a way to avoid immune attack, and therefore, linking RIPK3-induced necroptosis to the pathway ensures that cells infected by such viruses still die. Another, similar argument relates to c-FLIPL, which is rapidly turned over in cells: When intracellular parasites (including viruses) disrupt the translation of cellular proteins, levels of c-FLIPL decline, thereby sensitizing the cell to necroptosis. The destruction of the cell not only limits the replication of the parasite, but also engages the inflammatory response elicited by the necrotic cells.
Because necroptosis appears to be a bona fide form of cellular suicide, it is possible that this cell death process can function in tumor suppression. As more is learned about this phenomenon, cancers may be found in which oncogenesis is offset by engagement of necroptosis. Alternatively, there may be ways to exploit the process for tumor cell destruction in some forms of cancers.
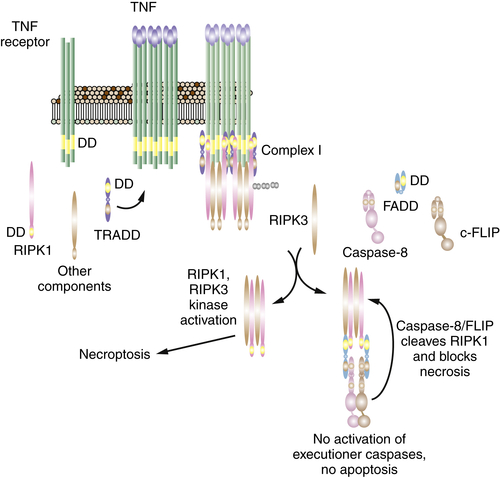
Figure 14-7 The necroptosis pathway See text for explanation.
Necroptosis, RIP Kinases, and Cancer Therapy
One area of cancer therapy involves a class of compounds called Smac mimetics, as they were originally designed to mimic the effects of the IAP antagonists (such as Smac) released upon MOMP. In fact, we now know that these agents work through targeting the cellular IAP molecules, c-IAP1 and c-IAP2.
The c-IAP molecules are E3 ligases that have, among their targets, the kinases RIPK1 and RIPK3. In healthy cells, the c-IAPs restrict the accumulation of these kinases and therefore prevent their activation. When cells are exposed to the Smac mimetics, the kinases accumulate, but if c-FLIPL is present, the action of FADD-caspase-8–c-FLIPL prevents death. However, if c-FLIPL is absent, the cells will die by caspase-8–mediated apoptosis or via necroptosis.
Autophagy
Autophagy, or cellular self-eating, is a process cells use to collect intracellular proteins, cytoplasm and organelles, and deliver them to lysosomes where they are degraded and recycled. 74 There are multiple forms of autophagy that use different routes to deliver cargo to lysosomes for degradation. 74 We focus here on macroautophagy (autophagy hereafter), where double membrane vesicles called autophagosomes capture cargo and then fuse with lysosomes. Autophagy has a dual role in cancer: It can be tumor suppressive by preserving cellular and tissue health and can be tumor promoting by supporting mitochondrial metabolism and survival in stress. We discuss the contexts in which autophagy influences cancer development, progression, and response to therapy.
Autophagy was discovered more than 50 years ago and was found to be activated by starvation in yeast, which enabled identification and characterization of many of what would become known as the approximately 30 autophagy-related (atg) genes. 75,76 This helped establish the framework for autophagy regulation, autophagosome formation, and the capture, delivery, and degradation of cargo in lysosomes. Discovery of orthologs of atg genes in other model organisms and in humans broadened our understanding of the role of autophagy in health and in disease.
There are two major functions of autophagy. The first is to eliminate damaged or superfluous proteins and organelles, commonly referred to as the protein and organelle quality control function of autophagy. The second is to degrade and recycle intracellular components to sustain metabolism and homeostasis in the absence of external nutrients, commonly referred to as the catabolic function of autophagy. Autophagy occurs at a low basal level in normal cells and tissues and is dramatically upregulated by stress and starvation, which is critical for cellular and mammalian survival and homeostasis. 77 Variations of autophagy can also remodel tissues in development or capture and degrade intracellular pathogens and contribute to host defense. 74 Autophagy has been paradoxically referred to as type II programmed cell death, where instead autophagy is predominantly a survival mechanism. The long-ago observations of autophagosomes in dying cells that led to this designation are instead likely a result of cells attempting to save themselves by activating autophagy. 78
Protein and Organelle Quality Control
A common feature of cells and tissues with defective autophagy is the accumulation of aggregated proteins and dysfunctional mitochondria and other organelles, which can be toxic. 79,80 Autophagy is specifically critical to purge bad mitochondria on a regular basis; otherwise cells fill up with these defective organelles. Cells can degrade individual soluble proteins via the ubiquitin-proteasome pathway; however, autophagy is the only mechanism cells have for large-scale bulk degradation and for degrading large structures such as protein aggregates and entire organelles. Autophagy is thereby a nonredundant process critical for cellular physiology. Tissues from mice deficient for essential autophagy (atg) genes accumulate autophagy substrates including lipids, aggregated proteins, and abnormal organelles, particularly mitochondria. Autophagy deficiency causes neurodegeneration, 81,82 liver damage and inflammation, 83–86 muscle cell deterioration, 87 degeneration of pancreatic islets, 88,89 and impairment of lymphocyte homeostasis. 90 Liver and brain appear to require autophagy more than other tissues.
Cell Catabolism
A common function of autophagy that is conserved from yeast to mammals is to provide survival in starvation through intracellular recycling. 91–94 This is dramatically demonstrated by the observation that mice deficient for the essential autophagy gene atg5 are born but fail to survive the neonatal starvation period, and their tissues display signs of energy impairment. 92 In the absence of external nutrients, catabolic recycling of proteins and organelles by autophagy is essential to sustain mammalian metabolism and survival.
Quality Control and Catabolism Overlap
Mitochondria are the nexus of functional overlap between the quality control and catabolic functions of autophagy. On the one hand, the quality control activity of autophagy is required to preserve the functioning pool of mitochondria to sustain their metabolic activity and cell survival. On the other hand, the catabolic activity of autophagy supplies substrates to mitochondria for them to metabolize. Thus, a major role for autophagy is in the regulation of mitochondrial metabolism, which affects cellular energy status, oxidative stress, signaling, and anabolism.
Dual and Context-Specific Role for Autophagy in Cancer
Both the quality control and catabolic functions of autophagy promote cell, tissue, and organismal homeostasis and survival, and in most circumstances this suppresses or delays cell death mechanisms of apoptosis and necrosis by promoting cellular health. 95,96 Autophagy has a dual and context-specific role in cancer. 97,98 By preserving cell and tissue health in some circumstances, autophagy can be tumor suppressive. Autophagy, however, can enable survival of cancer cells (and normal cells) in stress and in response to activation of some oncogenic pathways, and in this context autophagy can be tumor promoting.
Process of Autophagy
The central functions of autophagy have at their core the ability to identify and capture cargo and deliver it to the lysosomal compartment where it is degraded. 74 The breakdown products of intracellular proteins and organelles are then released from lysosomes into the cytoplasm, where they can be reutilized. 93 These amino acids, nucleosides, sugars, and fatty acids are used either as substrates to drive metabolic pathways or as building blocks for generation of new biomass. Regulators of autophagy ensure that the right cargo is degraded at the right time in the right cells.
In normal conditions, autophagy functions at low basal levels to ensure the occasional, selective, and necessary turnover of proteins and organelles due to aging and normal wear and tear. Damaged proteins and organelles display the “eat me” signal, the addition of polyubiquitin chains to proteins, which are recognized by autophagy receptors that recruit autophagosomes. In response to stress and starvation, autophagy is robustly induced. This ensures the elimination of damaged proteins and organelles that are induced by stressful conditions and also provides an alternate source of bulk metabolic substrates through intracellular recycling when external nutrient sources are limiting.
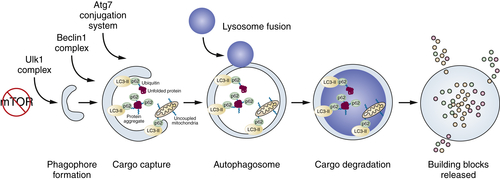
Figure 14-8 Process of autophagy See text for explanation.
There are many growth regulatory and stress pathways that control the switch from basal to induced autophagy. Many of these pathways converge on the major nutrient-sensing and cell growth–promoting mammalian target of rapamycin (mTOR). Nutrients activate mTOR, which shuts off initiation of autophagosome formation by directly phosphorylating components of the atg1/unc-51–like kinase 1 (ULK1) complex (see Figure 14-8). 99 Starvation suppresses mTOR activity, de-repressing autophagy and providing a direct link between nutrient availability, cell growth, and catabolism (Figure 14-8 ). Simply put, cells have no need to eat themselves when there is plenty of food, and regulation of autophagy by mTOR integrates that important relationship.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


