Figure 13-1 Mitochondria efficiently degrade metabolic fuels to generate ATP Metabolic fuels—carbohydrates, fats, and proteins—are broken down into their constituent units: glucose, fatty acids, and amino acids. These molecules are further degraded into metabolic intermediates that enter the tricarboxylic acid (TCA) cycle. Glucose-derived pyruvate, fatty acids, and many amino acids enter the TCA as acetyl-CoA, which condenses with oxaloacetate to form citrate. A series of biochemical reactions oxidize citrate back to oxaloacetate, while using the high-energy electrons released by these oxidation reactions to reduce the electron carriers NAD+ and FAD to NADH and FADH2, respectively. NADH and FADH2 carry these electrons to complex I and complex II of the electron transport chain, regenerating NAD+ and FAD. The high-energy electrons are passed through the electron transport chain (complex I-III-IV or complex II-III-IV), and their energy is used to pump protons from the mitochondrial matrix into the inner membrane space, generating an electrochemical gradient known as the mitochondrial membrane potential. Ultimately, the electrons reduce molecular oxygen to form water. The energy released by protons flowing down their electrochemical gradient through complex V/ATP synthase is harnessed to synthesize ATP from ADP and Pi. Reactive oxygen species (ROS) are formed when electrons leak out of the electron transport chain prematurely, combining with oxygen to form the superoxide radical (O2 .− ). Although ROS is an inevitable by-product of oxidative phosphorylation, its production can be increased whenever flow through the electron transport chain is slowed, whether by heightened membrane potential or reduced oxygen concentrations. Metabolites are shown in blue. Electron carriers are shown in green.
Regulation of Cell Metabolism
During conditions of nutrient scarcity, anabolic programs are suppressed in favor of energy-generating catabolic pathways. Conversely, multiple cellular metabolic pathways must be coordinately rewired to support cell proliferation. Elevated nutrient uptake—particularly of glucose and glutamine—provides the substrates for cell growth. Rapid catabolism of glucose via glycolysis produces sufficient ATP and NADPH to support energy-dependent anabolic reactions while generating the metabolic intermediates that will be critical for macromolecular biosynthesis. Similarly, catabolism of glutamine will help maintain bioenergetics while ensuring adequate substrates for cell growth. Multiple regulatory mechanisms ensure that energy production and macromolecular biosynthesis are appropriately balanced with the metabolic needs of the cell.
Unicellular organisms are directly exposed to the environment and any fluctuations in nutrient availability that may occur. Consequently, these organisms have elegant mechanisms to sense the available nutrients and rewire their metabolism accordingly. Thus, for unicellular organisms, nutrient availability directly controls the signals that regulate growth and proliferation. If a unicellular organism is in an environment with abundant nutrients, these nutrients will directly activate signaling pathways that instruct the cell to engage anabolic metabolic pathways and to undergo cell division. Conversely, conditions of low nutrient availability will halt cell growth and division. Cells will engage catabolic pathways in order to produce energy to survive through the period of scarcity (Figure 13-2 ).
In contrast, metazoans have complex organ systems that maintain a relatively constant level of extracellular nutrients throughout the body. Cell-autonomous metabolic regulation would be catastrophic, as well-fed cells might undergo aberrant growth and proliferation. Thus, nutrient availability alone cannot determine whether cells engage anabolic or catabolic pathways; the metabolism of individual cells must be aligned with the needs of the organism as a whole. This coordination is largely achieved through extracellular growth factors that regulate nutrient uptake and utilization. Binding to receptors on the cell surface, growth factors stimulate intracellular signal-transduction cascades that regulate many facets of nutrient uptake and metabolism. In particular, growth factor signaling enables cells to take up high levels of nutrients such as glucose and glutamine and to engage in anabolic pathways supporting cell growth (see Figure 13-2). 9 In this manner, systemic signals can target individual cells (expressing the proper growth factor receptor) to induce specific activities, thereby ensuring that the behavior of individual cells is tailored to the needs of the entire organism. In the absence of growth factor signaling, metazoan cells are largely quiescent, maintaining homeostasis by the efficient degradation of the limited nutrients they are directed to take up. By engaging oxidative, catabolic pathways that preserve intracellular energy levels, these cells are able to survive and fulfill their allotted functions.
Most cells have the capacity to engage both anabolic and catabolic pathways, and the balance between the two—the balance that guards against inappropriate cell proliferation while maintaining cell survival—is carefully regulated by a number of factors. External signals such as growth factors instruct the cell to grow, activating anabolic reactions and driving nutrient uptake accordingly. The activities of key metabolic enzymes that determine whether metabolites enter anabolic or catabolic pathways are regulated by posttranslational modifications, most commonly phosphorylation triggered by growth factor signaling. Metabolites themselves function as critical allosteric regulators of metabolic enzyme activity, increasing or inhibiting flux through metabolic pathways according to the needs of the cell. For example, signals of abundant energy stores such as ATP and NADH can allosterically inhibit multiple enzymes that otherwise promote the channeling of metabolites into catabolic, energy-producing pathways. This allows for the diversion of metabolites toward anabolic processes when energy supply is plentiful. Similarly, both metabolic cues and growth factor signaling pathways can regulate the expression levels of metabolic enzymes. Through these mechanisms, cells carefully control the activation of anabolic or catabolic metabolic programs and ensure that during cell proliferation, metabolic pathways are coordinately rewired to support cell growth.
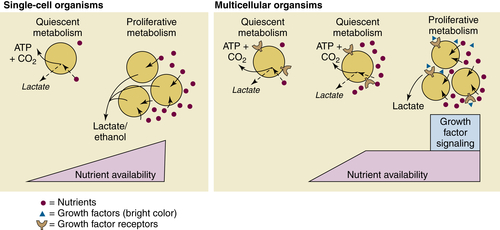
Figure 13-2 Proliferative metabolism is differentially regulated in single-cellular and multicellular organisms Quiescent cells in both single-cellular organisms and multicellular organisms engage in oxidative metabolism, efficiently converting available nutrients to ATP and carbon dioxide. In the presence of abundant nutrients, single-cellular organisms will engage in proliferative metabolism, consuming high levels of nutrients to produce biomass while excreting large amounts of lactate or other anaerobic fermentation products, such as ethanol. In contrast, the presence of abundant nutrients is insufficient to trigger proliferative metabolism in multicellular organisms, which require additional growth factor signals in order to engage proliferative metabolism. Once stimulated, these cells will undergo high rates of glycolysis to fuel cell growth.
A major hallmark of cancer is the development of cell-autonomous regulation of cell growth. 10 Through multiple genetic events, including activation of growth factor signal transduction pathways (oncogenes) and loss of inhibitory signals (tumor suppressors), cancer cells circumvent dependence on external growth factor signaling. Constitutive activation of growth factor signaling pathways ensures that cancer cells are not subject to the normal regulation of metazoan cells. Consequently, cancer cells exhibit metabolic transformation, taking up high levels of nutrients and engaging in proliferative metabolism to support unchecked cell growth.
The Metabolic Profile of Cancer Cells
It is increasingly clear that cancer cells exhibit metabolic phenotypes that are similar to rapidly proliferating normal cells, with the major difference that the metabolic alterations in cancer cells stem from oncogenic cell-autonomous signaling, rather than the appropriate result of specific growth signals originating outside the cell. The characteristic metabolic features of rapidly proliferating tumor cells include elevated glucose uptake and glycolysis, increased glutamine uptake and utilization, and enhanced lipid and nucleotide biosynthesis. This section discusses the common metabolic signatures of tumor cells and how these metabolic alterations may support tumor growth.
Aerobic Glycolysis: The Warburg Effect
The most well-known and prevalent metabolic change associated with cancer cells is the enhanced uptake and metabolism of glucose, often referred to as the Warburg effect. In 1926, Otto Warburg noted that rapidly proliferating ascites cancer cells take up high levels of glucose and produce large amounts of lactate, even in the presence of oxygen. This finding was not intuitive: Work begun by Louis Pasteur had shown a clear inverse relationship between oxygen availability and the rate of glucose fermentation to lactate. The ability of eukaryotic cells to switch between anaerobic energy production through fermentation to aerobic energy production through oxidative phosphorylation depending on the presence of oxygen is known as the Pasteur effect (Figure 13-3 ). In cancer cells in the presence of oxygen, one might expect glucose to be metabolized to pyruvate, which would then be completely oxidized in the mitochondrion to produce ATP through the oxygen-dependent process of oxidative phosphorylation (see Figure 13-1). The surprising observation that cancer cells converted pyruvate to lactate despite abundant oxygen availability—the process of aerobic glycolysis—led Warburg to speculate that mitochondrial function is impaired in tumor cells, forcing a reliance on glycolytic metabolism. 11
We now know that most cancers do not exhibit impaired mitochondrial energy production. 12 Moreover, research increasingly suggests that mitochondrial metabolic pathways are not simply catabolic and energy-producing; they may also play a critical role supporting anabolic biosynthetic pathways, as discussed later. These findings indicate that high aerobic glycolysis is not the secondary result of a metabolic failure, but rather a specific adaptation that promotes cell growth. Despite the centrality of the Warburg effect to cancer cell metabolism, there is still some debate as to how aerobic glycolysis confers a proliferative advantage to tumor cells.
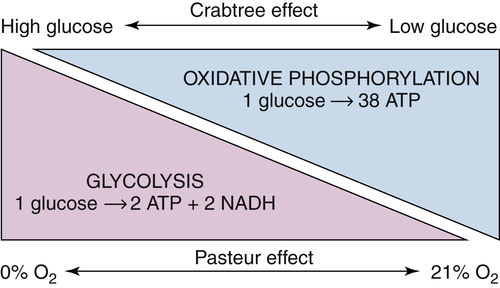
Figure 13-3 Normal, nonproliferating cells coordinate the balance between glycolysis and oxidative phosphorylation based on oxygen and glucose availability The Crabtree effect describes the inverse relationship between glucose availability and oxidative phosphorylation. In normal cells, abundant glucose promotes glycolytic metabolism and inhibits oxidative metabolism; conversely, low glucose availability stimulates mitochondrial oxidative phosphorylation to maximize ATP production from the available glucose. Similarly, the Pasteur effect summarizes the influence of oxygen tension on cellular metabolism: as cells adapt to low oxygen, they reduce their reliance on oxygen-dependent oxidative phosphorylation and increase their glycolytic rate to compensate. The Warburg effect appears to contradict these relationships, as highly proliferative cells undergo glycolytic metabolism even in the presence of abundant oxygen.
The Paradox of the Warburg Effect
Glycolysis comprises a series of reactions that convert 1 molecule of glucose to 2 molecules of pyruvate, generating 2 molecules each of ATP and NADH (Figure 13-4 ). If oxygen is present, pyruvate is converted to acetyl-CoA in the mitochondrion, and acetyl-CoA is oxidized by the TCA cycle, producing 1 molecule of GTP and four pairs of high-energy electrons that will be used to fuel OXPHOS (3 molecules of NADH and 1 molecule of FADH2). As the glycolytic reactions occur in the cytosol, the reducing equivalents of the NADH generated by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) must be transferred to the mitochondrion in order for their energy to be harnessed into ATP. The malate-aspartate shuttle and the glycerol-3-phosphate shuttle transfer the electrons into the mitochondrion for oxidation by the electron transport chain. All together, this process of complete glucose oxidation produces 38 molecules of ATP. Thus, when oxygen is present, glucose can be efficiently converted to ATP while regenerating the NAD+ required to maintain glycolysis.
If NADH is not oxidized to NAD+, the subsequent depletion of NAD+ will inhibit GAPDH and block glycolysis. To avoid this, under anaerobic conditions lactate dehydrogenase (LDH) reduces pyruvate to lactate, consuming NADH and producing NAD+. In total, therefore, anaerobic glycolysis yields two molecules of ATP and two molecules of lactate that are secreted from the cell.
The paradox of the Warburg effect is that cells convert pyruvate to lactate even in the presence of oxygen. If glucose oxidation produces 19 times more ATP than anaerobic glycolysis, what benefit could a cell gain from choosing the less efficient route? And why would a cell that is growing rapidly throw away valuable carbon units in the form of lactate? Warburg’s studies described an astonishing rate of lactate production in ascites tumor cells—cells produced up to 30% of their dry weight in lactic acid per hour. 13 Although it is now known that this is a much higher rate than for most tumors, it is clear that aerobic glycolysis occurs at a high rate in many tumor cells. There are several potential explanations for how tumors could sustain such apparently wasteful metabolism and the benefits that rapid aerobic glycolysis might bestow:
1. Aerobic glycolysis is a source of rapid ATP generation. According to Warburg’s calculations, the high rate of glucose consumption enabled cells to produce approximately the same amount of ATP through fermentation as through respiration. 11 Similarly, a series of studies demonstrated that stimulating cells to proliferate increased ATP turnover, as expected; however, the increased ATP demand was met entirely by increased glycolytic flux and not by any increase in ATP production by oxidative phosphorylation. 14 Thus, the relative inefficiency of ATP production may be counterbalanced by the high rate of glucose consumption. Furthermore, tumor cells may extract more than two molecules of ATP per molecule of glucose: Several studies demonstrate that cytosolic-mitochondrial NADH shuttles are active in tumor cells, indicating that tumors can oxidize GAPDH-derived NADH in the mitochondrion to produce ATP. 15–18 In addition, glycolysis generates cytosolic ATP very rapidly: The conversion of glucose to lactate can take mere seconds. This could benefit tumor cells undergoing high rates of protein, lipid, and nucleotide biosynthesis. Thus, as long as glucose supplies are not limited, high levels of glycolysis may provide an advantageously rapid and plentiful source of ATP.
2. Adaptation to hypoxia. Cells in solid tumors experience notoriously harsh and varied metabolic conditions as tumor growth disrupts the normal tissue architecture. Blood vessels inside the tumors are often dysfunctional or nonexistent. Consequently, tumor cells commonly experience periods of intermittent hypoxia. Survival in hypoxia requires metabolic adaptations, including increasing glycolysis and downregulating mitochondrial fuel oxidation. 19 A cell already primed with these metabolic adaptations would be more likely to survive inside a solid tumor. Indeed, cells cultured ex vivo from solid tumors often display high lactate production even when well oxygenated, suggesting that elevated glycolysis is a fundamental feature that either predisposes cells toward tumor formation or is selected for early in tumor development. It is worth noting that because hypoxia typically results from inadequate blood supply, cells rarely experience hypoxia without concomitant deprivation of nutrients such as glucose that are also provided by the vasculature. Thus, although glycolytic metabolism may increase the likelihood of survival during periods of intermittent hypoxia, sustained hypoxia will likely negatively influence cell growth regardless of metabolic adaptations.
3. Acidification of microenvironment. There is some evidence that the apparently wasteful secretion of lactate may itself confer a selective advantage to tumor development. Cells export lactate through the family of H+-linked monocarboxylate transporters. By acidifying the microenvironment, lactate export may promote the death of normal cells and extracellular matrix degradation to enhance tumor invasion. 20 In support of this model, studies suggest that acidic conditions can stimulate tumor cell invasion in vitro, and in vivo interventions to increase pH can reduce spontaneous metastases. 21 Intriguingly, some studies suggest that lactate may also be used as a fuel source. Lactate exported from hypoxic cells can be used as a substrate for oxidative metabolism in normoxic cells within the same tumor, preserving glucose for the hypoxic cells while providing fuel for the normoxic cells. 22 Thus, lactate production may not be an unfortunately costly by-product of glycolytic metabolism, but may also serve pro-tumorigenic roles. Indeed, blocking conversion of pyruvate to lactate by suppressing LDH expression can impair tumorigenesis. 23 Clearly, the production of lactate is critical for cancer cell growth, whether by maintaining oxidized NAD+ to promote glycolysis, acidifying tumor surroundings to promote cancer cell survival, or providing a means for efficient substrate allocation in a metabolically diverse microenvironment.
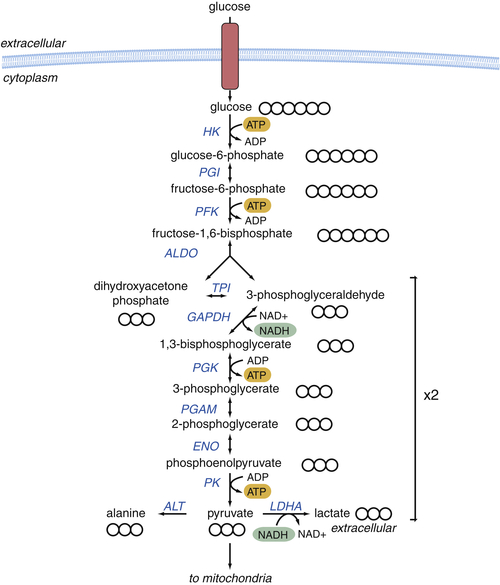
Figure 13-4 Glycolysis produces 2 ATP and 2 NADH for each molecule of glucose Glucose is metabolized to pyruvate through a series of biochemical reactions in the cytosol of the cell. This process yields a net of 2 ATP and 2 NADH molecules for each molecule of glucose. Pyruvate can be converted to lactate to regenerate the NAD+ required to maintain flux through glycolysis, transaminated to alanine, or transported to the mitochondrion for further oxidation. Circles represent the number of carbons in each metabolite. Enzymes are shown in blue; reduced NADH is shown in green; ATP is shown in orange. ALDO, aldolase; ALT, alanine transaminase; ENO, enolase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HK, hexokinase; LDHA, lactate dehydrogenase; PFK, phosphofructokinase; PGAM, phosphoglycerate mutase; PGI, phosphoglucose isomerase; PGK, phosphoglycerate kinase; PK, pyruvate kinase; TPI, triosephosphate isomerase.
Glycolysis Provides Key Intermediates to Support Cell Growth
The preceding examples provide clues for how rapid aerobic glycolysis might provide a growth or survival advantage to tumor cells. However, we now know that aerobic glycolysis as Warburg described it—the conversion of glucose to lactate—is not the exclusive fate of glucose in cancer cells. Glucose is a major source of many of the building blocks of macromolecular biosynthesis, and it is clear that these various anabolic fates of glucose are an important component of the elevated glycolysis in cancer cells.
Glucose-Derived Metabolites Contribute to All Classes of Macromolecules
Glycolytic flux provides substrates for biosynthetic reactions that are required for the synthesis of lipids, nucleotides, and proteins (Figure 13-5 ). Glycolytic intermediates can be diverted to the pentose phosphate pathway to produce ribose 5-phosphate, which serves as the foundation for the de novo synthesis of purines and pyrimidines. The oxidative arm of the pentose phosphate pathway also generates NADPH, which can provide reducing equivalents for nucleotide and fatty acid synthesis. Similarly, the glycerol required for phospholipid synthesis is derived from the glycolytic intermediate dihydroxyacetone phosphate. As the major component of mammalian cell membranes, phospholipid synthesis is critical for cell growth. Glycolytic intermediates can also contribute to amino acid synthesis: 3-Phosphoglycerate can be converted to serine and ultimately glycine.
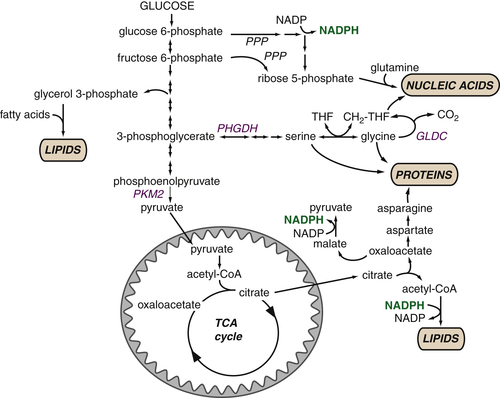
Figure 13-5 Glucose metabolism provides critical intermediates for cell growth Glycolytic intermediates provide the substrates for the pentose phosphate pathway (PPP) that generates the ribose 5-phosphate that is critical for nucleotide biosynthesis. The oxidative arm of the PPP, which utilizes glucose 6-phosphate, additionally generates NADPH. Glycerol 3-phosphate, which contributes the glycerol head groups for phospholipid biosynthesis, is formed from the intermediate dihydroxyacetone phosphate. 3-Phosphoglycerate provides the foundation for the serine synthesis pathway, which can further fuel glycine production for protein synthesis. This pathway also contributes to the pool of one-carbon units (CH2-THF) that are used in nucleotide biosynthesis. Glucose-derived citrate provides the acetyl-CoA that represents the fundamental building block for the synthesis of the fatty acids that comprise cellular lipids. Key metabolic enzymes are shown in blue; NADPH required for biosynthetic reactions is shown in green. CH 2 -THF, methylene tetrahydrofolate; GLDC, glycine decarboxylase; PHGDH, phosphoglycerate dehydrogenase; PKM2, pyruvate kinase M2 isoform; PPP, pentose phosphate pathway; THF, tetrahydrofolate.
A series of recent studies documenting tumors whose growth depends on diversion of glycolytic flux to anabolic pathways lends support to the hypothesis that glycolytic intermediates play a key role fueling tumor growth. Phosphoglycerate dehydrogenase (PHGDH), the enzyme that catalyzes the first step of the serine synthesis pathway that branches from glycolysis, is frequently amplified in melanoma and breast cancers, and flux into the serine synthesis pathway can support tumor cell growth. 24,25 Similarly, multiple tumor types display high levels of glycine decarboxylase (GLDC), which generates methylene tetrahydrofolate necessary for pyrimidine synthesis, and GLDC expression can promote cellular transformation and tumorigenesis. 26
Macromolecular Synthesis, Not ATP, May Be the Major Purpose of Elevated Glycolysis in Cancer Cells
The importance of glycolytic intermediates as biosynthetic building blocks supporting cell proliferation was underscored by the observation that most proliferating cells express the M2 isoform of pyruvate kinase (PK), whereas quiescent cells express the PKM1 isoform. 27 Pyruvate kinase catalyzes the final ATP-producing step of glycolysis, converting phosphoenolpyruvate to pyruvate. Perhaps counterintuitively, PKM2 has less intrinsic activity than PKM1; PKM2 is also inhibited by growth factor signaling. These observations suggest that inhibiting the final step of glycolysis can be beneficial for cell growth. By slowing the final step of glycolysis, PKM2 enables accumulation of upstream glycolytic intermediates that can then be diverted into biosynthetic pathways. These findings indicate that proliferating cells do not undergo glycolysis for the sole purpose of ATP production; rather, they appear to selectively slow pyruvate production in order to maximize the flow of glucose carbons into anabolic pathways.
Expression of PKM2 may have the added benefit of reducing ATP production. Although ATP is required for anabolic reactions, NADPH is quantitatively more important. Biosynthesis of many macromolecules requires more reducing equivalents than energy from ATP. 1 Consequently, if all glucose were oxidized completely, ATP production would far outstrip NADPH production, thus greatly circumscribing potential cell growth. Furthermore, ATP is a potent inhibitor of phosphofructokinase-1, a key enzyme in the regulation of glycolysis, and so ATP produced in excess of biosynthetic demands would have the adverse effect of blocking glycolysis, further reducing NADPH production.
Although cancer cells preferentially reduce pyruvate kinase activity, the final step in glycolysis cannot be abrogated completely, as pyruvate fulfills many important metabolic roles. The pyruvate produced at this step has three fates: It can be transaminated to alanine, reduced to lactate to regenerate NAD+, or imported to the mitochondrion, where it is converted to acetyl-CoA that can enter the TCA cycle. In many tumors, glucose consumption is high enough to support anabolic pathways while also maintaining pyruvate flux into the mitochondria and still resulting in high amounts of lactate secretion. It is likely that reducing pyruvate kinase activity slows glucose flux through glycolysis in order to increase the likelihood that glucose can be diverted into anabolic pathways rather than being completely and rapidly exported as lactate or oxidized in the mitochondria. Given the high rates of lactate produced from glucose (as much as 90% of glucose is converted to lactate and alanine in glioblastoma cells), 28 cancer cells still have appreciable pyruvate consumption. It is likely that despite the high percentage of pyruvate that is secreted as lactate, the rate of glycolysis is so high that sufficient pyruvate remains to fuel the TCA cycle.
The TCA Cycle as a Biosynthetic Hub
Just as glycolytic intermediates are harnessed to support anabolic pathways, proliferating cells rewire the TCA cycle to prioritize cell growth over ATP generation. In its traditional form, the TCA cycle begins with the condensation of pyruvate-derived acetyl-CoA with oxaloacetate to form citrate (Figure 13-6 ). A series of reactions in the mitochondrial matrix convert citrate back to oxaloacetate, releasing two carbons as two molecules of carbon dioxide and producing three molecules of NADH and one molecule of FADH2. NAD+ and FAD are regenerated when NADH and FADH2 donate their electrons to support oxidative phosphorylation. The cycle can continue by combining the recycled oxaloacetate with a new molecule of acetyl-CoA. In this manner, the oxidation of acetyl-CoA is used to maximize ATP production.
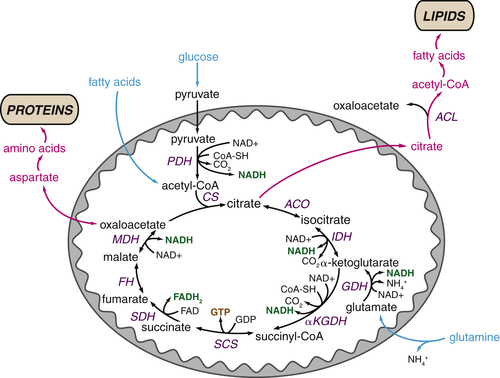
Figure 13-6 The tricarboxylic acid (TCA) cycle supports macromolecular biosynthesis Pyruvate derived from glucose enters the mitochondrion and is converted to acetyl-CoA by pyruvate dehydrogenase (PDH), and fatty acids are oxidized to produce acetyl-CoA units within the mitochondrion. Acetyl-CoA condenses with oxaloacetate to form citrate, which proceeds through the TCA cycle in highly oxidative tissues, releasing two carbons as carbon dioxide and regenerating oxaloacetate for further cycling. These reactions maximize ATP production from acetyl-CoA by reducing the electron carriers NADH and FADH2 to fuel the electron transport chain. In proliferating cells, citrate is also exported from the mitochondrion to the cytosol, where it can provide the acetyl-CoA required for the synthesis of fatty acids and cholesterol. Similarly, oxaloacetate can form aspartate and other amino acids, contributing to intracellular amino acid pools for protein synthesis. Both of these reactions deplete TCA cycle intermediates; consequently, cells must replenish TCA cycle metabolites in order to regenerate the oxaloacetate necessary for citrate synthesis. Most cells use glutamine to fulfill this anaplerotic role: glutamine is converted to glutamate and then α-ketoglutarate, preserving TCA cycle flux. Metabolites that fuel the TCA cycle are shown in light blue. Metabolite efflux from the TCA cycle is shown in pink. Enzymes are shown in dark blue, reduced electron carriers in green, and high-energy triphosphates in orange. PDH, pyruvate dehydrogenase; ACL, ATP-citrate lyase; ACO, aconitase; CoA-SH, coenzyme A; CS, citrate synthase; FH, fumarate hydratase; GDH, glutamine dehydrogenase; IDH, isocitrate dehydrogenase; αKGDH, α-ketoglutarate dehydrogenase; MDH, malate dehydrogenase; SCS, succinyl-CoA synthetase; SDH, succinate dehydrogenase.
TCA Cycle Metabolites Are Required for Macromolecular Biosynthesis
In proliferating cells, citrate is not only oxidized in the TCA cycle but also can be exported to the cytosol where it is converted back to oxaloacetate and acetyl-CoA by the enzyme ATP citrate lyase (ACL). Cytosolic acetyl-CoA provides the substrate for the synthesis of fatty acids, cholesterol, and prostaglandins (Figure 13-7 ). Through this pathway, glucose acts as the major substrate for de novo lipogenesis; consequently, ACL inhibition blocks cell proliferation and inhibits tumor growth. 28–30
Citrate is not the only TCA cycle metabolite that has an important biosynthetic role. Oxaloacetate and α-ketoglutarate can provide the carbon backbone for nonessential amino acids, which are used for protein and nucleic acid synthesis (see Figure 13-6). In this manner, multiple TCA cycle metabolites are diverted to other pathways that support cell growth.
Glutamine Plays Several Roles Supporting the TCA Cycle and Anabolic Metabolism
Although efflux of TCA cycle metabolites supports anabolic reactions, this results in depletion of TCA cycle intermediates from the mitochondrial matrix. Growing cells must draw on alternative sources to maintain the oxaloacetate required for citrate synthesis. Replenishment of TCA cycle intermediates, a process known as anaplerosis, is thus critical to maintain both a constant supply of metabolites for synthesis and appropriate ATP production through oxidative phosphorylation. Most proliferating cancer cells meet their anaplerotic needs through the catabolism of glutamine (see Figure 13-6). Glutamine is converted to the TCA cycle intermediate α-ketoglutarate through a metabolic pathway known as glutaminolysis. Tumor cells take up high levels of glutamine, often in great excess of any other amino acid, and studies using carbon labeling techniques have demonstrated that glutamine is the major source of carbon for the cellular oxaloacetate pool in cancer cell lines. 28
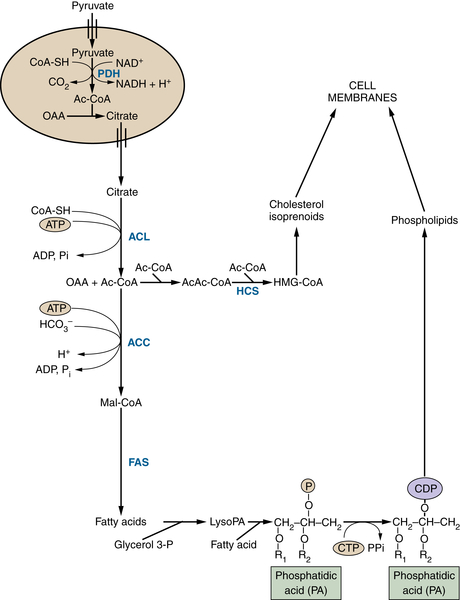
Figure 13-7 Glucose-derived acetyl-CoA fuels lipid and sterol synthesis Pyruvate derived from glycolysis can generate citrate in the mitochondrion. This citrate can be exported to the cytosol to provide acetyl-CoA for the synthesis of fatty acids, phospholipids, cholesterol, and isoprenoids, all of which are critical for membrane biogenesis and function. Metabolites are in black; enzymes are in blue. ACC, Acetyl-CoA carboxylase; Ac-CoA, acetyl-CoA; ACL, ATP-citrate lyase; CDP, cytidine diphosphate; CoA-SH, coenzyme A; CTP, cytidine triphosphate; FAS, fatty acid synthetase; glycerol-3-P, glycerol 3-phosphate; HCS, HMG-CoA synthetase; HMG-CoA, 3-hydroxy-3-methylglutaryl CoA; lyso PA, lysophosphatidic acid; Mal-CoA, malonyl-CoA; OAA, oxaloacetate; Pi, inorganic phosphate; R, acyl group on a lipid molecule.
By maintaining flux through the TCA cycle—and therefore delivery of electrons to the electron transport chain—glutamine plays a critical role maintaining mitochondrial bioenergetics in many cancer cells. Consequently, many cancer cell lines are absolutely dependent on glutamine for survival. Addition of cell-permeable TCA cycle analogs can rescue death of glutamine-deprived cells, highlighting the importance of glutamine as an anaplerotic substrate in cancer cells. 31,32
Proliferating cells rely on glutamine to fulfill additional biosynthetic roles. First, glutamine provides an important source of nitrogen for synthesis of nonessential amino acids and nucleotides. Glutamine is required for two independent steps in purine nucleotide synthesis, and oxaloacetate-derived aspartate is required for a third (Figure 13-8 ). Similarly, two steps of pyrimidine synthesis require glutamine. In all cases, glutamine donates nitrogen in the form of an amide group and is converted to glutamic acid, which provides a major source of nitrogen for amino acid synthesis. Transaminases can transfer the amine group from glutamic acid to α-ketoacids, which are themselves derived from the catabolism of glucose or glutamine, producing alanine, serine, aspartate, and ornithine. In turn, these amino acids act as precursors for the synthesis of glycine, cysteine, arginine, and asparagine. Likewise, two enzymatic reactions directly convert glutamate to proline. In this manner, glucose and glutamine contribute to the synthesis of every nonessential amino acid except for tyrosine, which is directly produced from the essential amino acid phenylalanine.
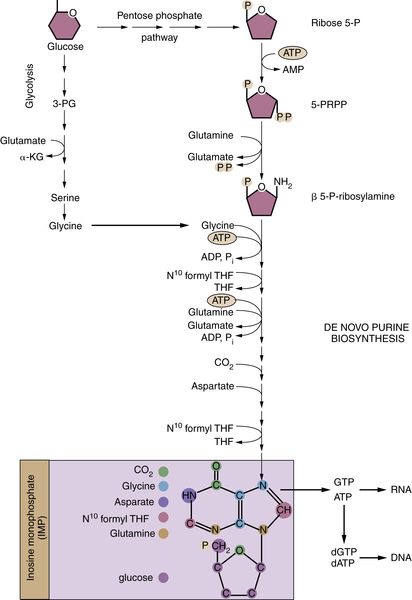
Figure 13-8 Nucleotide biosynthesis requires multiple metabolic inputs Nucleotide biosynthesis requires inputs from several metabolic pathways, highlighting why cells must coordinately regulate multiple metabolic pathways during proliferation. The de novo synthesis of purines (shown) and pyrimidines requires glucose, several amino acids, and one-carbon groups from folate metabolism. The origin of individual carbons and nitrogens on inosine monophosphate (IMP), the precursor to GTP and ATP, are color-coded in the purple box. α-KG, α-ketoglutarate; N10 formyl THF, N10 formyl tetrahydrofolate; 3-PG, 3-phosphoglycerate; β 5-P-ribosylamine, β 5-phosphate-ribosylamine; PRPP, 5-phosphoribosyl pyrophosphate; ribose 5-P, ribose 5-phosphate; P, phosphate group.
Intriguingly, cancer cells may convert up to 60% of glutamine carbon into lactic acid, an ostensibly wasteful secretion analogous to the Warburg effect. 28 One possible explanation for this behavior is that the conversion of glutamine to lactate produces NADPH, which is absolutely required for many anabolic reactions. NADPH is produced when malic enzyme converts glutamine-derived malate to pyruvate, which is subsequently reduced to lactate and secreted. In this manner, glutamine may contribute to all three needs of proliferating cells: Glutamine can provide the carbon and nitrogen for most metabolic building blocks, maintain ATP production to support bioenergetics, and generate reducing equivalents required for many anabolic reactions.
TCA Cycle Rearrangements Highlight the Role of TCA Cycle Metabolites as Biosynthetic Precursors
In proliferating cancer cells, the TCA cycle can behave more as a source of anabolic substrates rather than a bona fide cycle. A prime example of how the TCA cycle prioritizes biosynthetic reactions is the reductive carboxylation of α-ketoglutarate to isocitrate. During conditions of hypoxia or mitochondrial dysfunction, NADH accumulates from a combination of increased glycolysis and reduced oxidation of NADH by the electron transport chain. Depletion of oxidized NAD+ poses a bioenergetic challenge for the cell: Both the conversion of glucose-derived pyruvate to acetyl-CoA and the production of oxaloacetate through the enzymes of the TCA cycle require NAD+. Furthermore, during periods of acute hypoxia, pyruvate is converted to lactate at the expense of acetyl-CoA. Both of these events would strongly impair citrate production and thus cell growth. How, then, do hypoxic cells proliferate?
Several groups have demonstrated that under these conditions, the TCA cycle can partially function in reverse (for examples, see Refs. 33–35). Normally, isocitrate dehydrogenase (IDH) catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate, consuming NAD+ and producing NADH. When NAD+ is limiting, both the mitochondrial and cytosolic isoforms of IDH can function in reverse, carboxylating α-ketoglutarate to isocitrate and producing oxidized NADP+ (Figure 13-9 ). In this manner, cells can use glutamine-derived α-ketoglutarate to generate citrate, providing a mechanism to maintain anabolic reactions even under hypoxic conditions that characterize the tumor microenvironment.
Genetic Mechanisms Driving Cancer Cell Metabolism
Metabolic transformation is a common feature of tumorigenesis, occurring in many tumor types in diverse tissues. It is logical that metabolic transformation and oncogenic transformation would go hand in hand: As cancer cells acquire mutations that enable pathological proliferation, they must also acquire the means to support cell growth. Thus, many of the mutations that promote cancer growth also trigger metabolic reprogramming. This section discusses several common genetic oncogenic events that reprogram metabolism to support tumor growth.
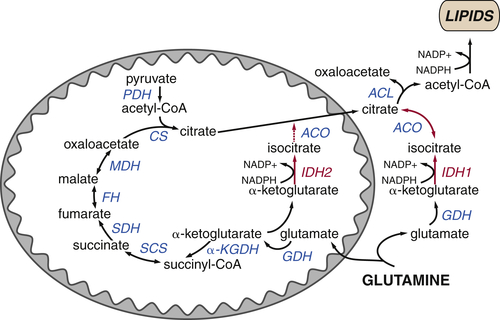
Figure 13-9 Reductive carboxylation supports lipid synthesis during hypoxia Citrate levels can be maintained even in cells with defective TCA cycles or during periods of hypoxia by the reductive metabolism of glutamine-derived α-ketoglutarate to citrate. Both mitochondrial and cytosolic isoforms of isocitrate dehydrogenase (IDH2 and IDH1, respectively) can function in reverse, reducing α-ketoglutarate to isocitrate and oxidizing NADPH to NADP+. Isocitrate is reversibly converted to citrate, which can be used to generate acetyl-CoA for lipid synthesis in the cytosol.
Activation of the PI3K Pathway Promotes Metabolic Transformation
In normal cells, extracellular cues regulate the intracellular signaling pathways that tightly coordinate metabolism and proliferation. In many cells, this coordination is accomplished by the phosphatidylinositol-3-kinase (PI3K) pathway, a highly conserved signaling pathway that responds to a variety of extracellular cues. When growth factors bind to their cell surface receptors, receptor tyrosine kinases (RTKs) activate PI3K, which phosphorylates phosphatidylinositol lipids at the plasma membrane. These phosphorylated lipids recruit and activate downstream effectors of the PI3K pathway, triggering an intracellular signaling cascade that promotes cell growth, survival, and metabolism.
Constitutive activation of PI3K provides a growth stimulus and is therefore a major mechanism of tumorigenesis. In normal cells, PI3K activity is tightly guarded by a number of factors, ensuring that pathway activity is set at appropriate levels. RTK activity is strictly controlled by growth factor availability. In addition, negative feedback loops prevent prolonged pathway activation: The lipid phosphatase PTEN dephosphorylates phosphatidylinositol species to dampen PI3K signaling.
Cancers exhibit a variety of mutations to circumvent this regulation and gain cell-autonomous activation of the PI3K pathway. 36 Amplifications or mutations of growth factor receptors occur in a variety of cancers. Common examples are the amplification of the Her2/Neu receptor in breast cancer and epithelial growth factor receptor (EGFR) in lung cancer. Similarly, deletion, downregulation, or loss of function of PTEN increases cancer susceptibility and promotes tumor progression. Several tumor types display activating mutations in the catalytic PI3K subunit, PIK3CA; likewise, amplification of PIK3CA and the major PI3K effector, Akt, are common oncogenic events. All together, genetic activation of the PI3K pathway is one of the most prevalent classes of tumorigenic mutations.
Whereas PI3K signaling influences numerous cellular functions, the serine/threonine kinase Akt and its effector, mechanistic target of rapamycin (mTOR), are the PI3K effectors most commonly implicated in tumorigenesis. Akt coordinates cell growth and survival in a large part by rewiring metabolism to promote nutrient uptake and biosynthetic activities. Certainly, with regard to the regulation of cell metabolism, Akt activation is likely the most important consequence of PI3K pathway activation.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


