Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .



Introduction
Cushing’s syndrome (CS) is an important cause of secondary hypertension. Hypertension is the most prevalent comorbidity and present in 75%–85% of adults and 47%–51% of children and adolescents with endogenous Cushing’s. In patients with ectopic CS, the prevalence of hypertension is 95% [ ]. Complex pathophysiological mechanisms are involved in the etiology of hypertension in patients with CS. Signs and symptoms of CS overlap with other conditions such as obesity, metabolic syndrome, and depression and thus it can take years to diagnose CS. Once there is clinical suspicion for CS, a complex testing algorithm including obtaining serum cortisol, salivary cortisol, urinary cortisol, and imaging studies are required to confirm the presence and to localize the disease source [ ], leading to further delay in the diagnosis and treatment. In fact, the mean time to diagnosis for patients with CS is 34 months (14 months for ectopic CS, 30 months for adrenal CS, and 38 months for pituitary CS) [ ]. Timely diagnosis is important to minimize morbidity and mortality, especially since there is evidence that duration of CS can impact outcome of surgery [ ]. Even after curative surgery, many of the sequelae of CS persist; hypertension is still present in more than 75% of such patients, perhaps due to irreversible vascular remodeling that might develop after prolonged glucocorticoid excess states [ ].
Moreover, hypertension can complicate the management of CS and the perioperative care of patients contemplating potentially curative surgical treatment. Therefore, it is imperative for clinicians to have a thorough understanding of the epidemiology, pathophysiology, clinical characteristics, management algorithms, and follow-up care of patients with CS-related hypertension. This chapter is an attempt to compile up-to-date evidence for this purpose.
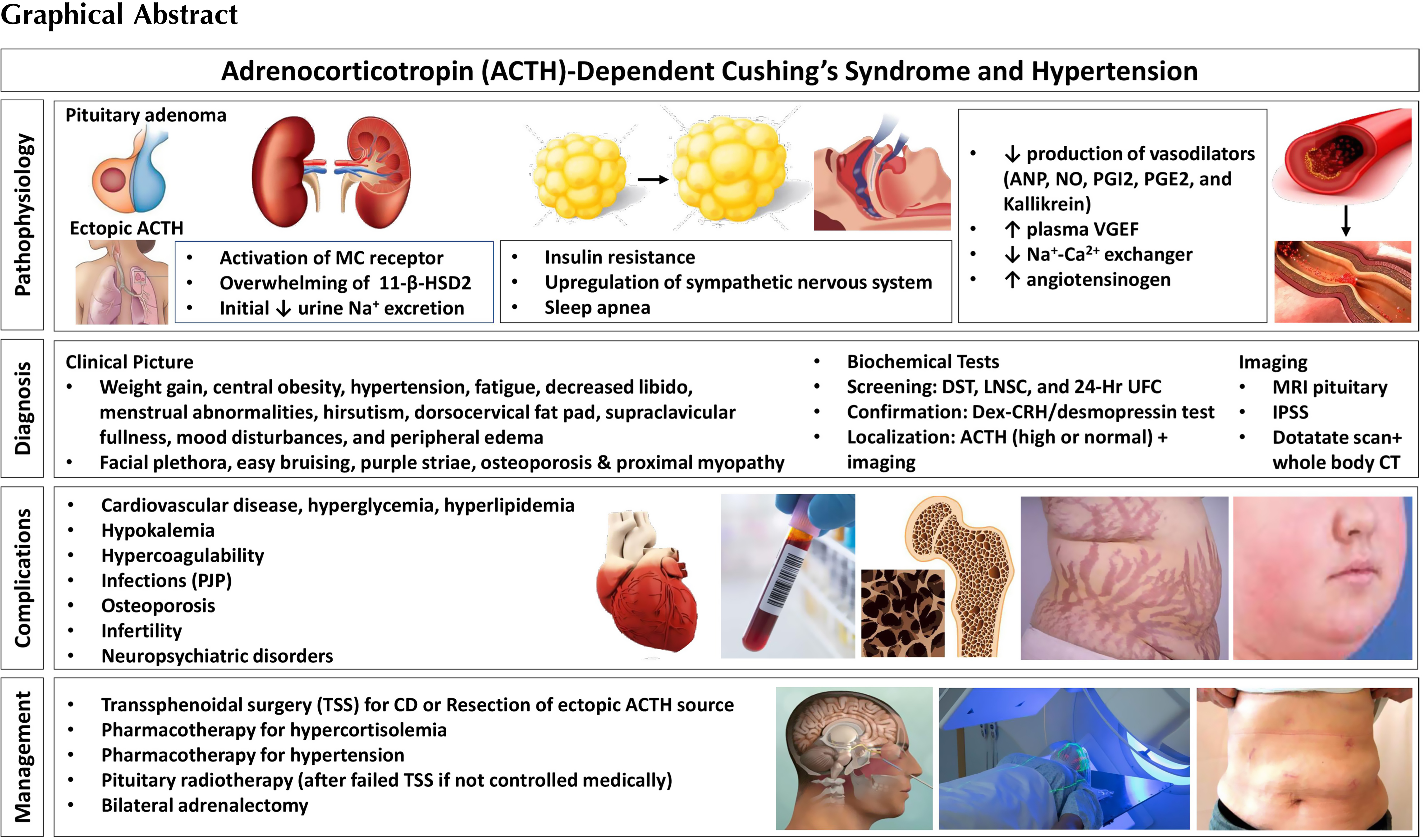
Clinical presentation, screening, and diagnosis of Cushing’s syndrome
Cushing’s syndrome is a severe medical condition caused by chronic exposure to elevated cortisol levels. Patients often present with weight gain, central obesity, hypertension, fatigue, decreased libido, menstrual abnormalities, hirsutism, dorsocervical fat pad, supraclavicular fullness, mood disturbances, and peripheral edema. However, these signs and symptoms are common in the general population, especially with the rise in global prevalence of obesity. Signs and symptoms with a higher specificity for CS include facial plethora, easy bruising, reddish purple striae >1 cm, osteoporosis, and proximal myopathy or muscle weakness [ ]. On laboratory work up, these patients may have hypokalemia (from excess cortisol acting on the mineralocorticoid (MC) receptor [ ]), hyperglycemia [ ], leukocytosis [ ], elevated liver enzymes [ ], secondary hyperparathyroidism [ ], and hyperandrogenism [ ].
Endogenous CS is caused by an adrenocorticotropin (ACTH)-secreting pituitary adenoma, cortisol secreting adrenal tumor, or an ectopic ACTH- or corticotrophin-releasing hormone (CRH)-producing tumor. Endogenous CS is rare and occurs in approximately 3/100,000 people [ ]. On the other hand, CS due to exogenous administration of glucocorticoids is much more common. Therefore, if there is suspicion of CS, the first step is to rule out exogenous glucocorticoid exposure (oral, injectable, inhaled, or topical preparations). Then cortisol excess should be confirmed with one or more of the following tests: 24-hour urinary free cortisol (UFC), late night salivary cortisol (LNSC), and/or dexamethasone suppression testing (DST) [ ]. UFC measures cortisol not bound to cortisol binding globulin (CBG). Urine collection should be done at least twice and collected along with urine creatinine and volume to ensure adequate collection. This test may not be accurate in those patients with chronic renal insufficiency [ ].
Patients with CS lose their normal circadian rhythm of cortisol production and overnight drop in cortisol levels and thus patients with CS have high late night salivary cortisol (LNSC) values. LSNC should be checked multiple times at bedtime or midnight [ ]. LSNC may not be accurate in smokers, shift workers or those with oral bleeding and infection [ ].
In healthy patients, 1 mg of dexamethasone should inhibit ACTH production and cortisol concentrations. A normal response to dexamethasone given at 11 p.m.–12 a.m. is cortisol suppression to <1.8 ug/dL. Dexamethasone levels should be checked as well to ensure that the patient does not have malabsorption, increased dexamethasone metabolism (caused by medications such as carbamazepine and phenytoin), decreased dexamethasone clearance (renal or liver insufficiency), or performed the test incorrectly. DST is recommended as the first-line screening test for CS in those with an adrenal incidentaloma. This test may not be accurate in patients with alterations in cortisol binding globulin and albumin, such as those on estrogen [ ]. Though these tests have high sensitivity, they do not have high specificity.
Physicians should use the results of several screening tests and clinical judgment to confirm the diagnosis of CS [ ]. Patients with severe obesity, psychiatric disorders, alcohol overuse, polycystic ovary syndrome, and some other metabolic disorders may have clinical features simulating CS and abnormal screening tests, known as non-neoplastic hypercortisolism or pseudo-Cushing’s. Special testing with dexamethasone-CRH test and/or desmopressin test are pursued to rule out pseudo-Cushing’s [ ].
Localization testing: distinguishing between ACTH-dependent and ACTH-independent Cushing’s syndrome
Once the diagnosis is confirmed, healthcare providers should determine if the CS is ACTH dependent (∼80%) or ACTH independent (∼20%) [ , ]. In patients with ACTH independent CS, ACTH is usually suppressed (<10 pg/mL), and adrenal imaging should be pursued to look for an adrenal adenoma or bilateral adrenal hyperplasia [ ]. However, an ACTH level of 10–20 pg/mL can also occur in those with adrenal CS. Raised dehydroepiandrosterone sulfate (DHEA-S) levels in the setting of low ACTH increase suspicion of adrenal carcinoma [ ]. For further reading on adrenal CS, please refer Chapter 14 .
In cases of ACTH dependence, ACTH level is high or normal (>20 pg/mL); differential diagnosis includes an ACTH producing pituitary adenoma (∼80%; Cushing’s Disease, CD) or ectopic ACTH secretion by a tumor (EAS; ∼20%) [ , ]. There is no single test that can easily distinguish between CD and ectopic CS. It is recommended to use a combination of imaging and diagnostic testing within the clinical context to determine the source of ACTH [ ]. Pituitary MRI should be obtained looking for a pituitary adenoma as it is the most common cause of ACTH-dependent CS. A finding of an adenoma >10 mm lends support to the diagnosis of CD [ ]. However, ∼12% of patients with EAS can also have abnormal pituitary imaging [ ]. On the other hand, normal pituitary MRI does not rule out pituitary source of CS because up to 40% of corticotroph adenomas are not visible on MRI [ ]. Inferior petrosal sinus sampling (IPSS) is recommended to localize the source of ACTH in cases of pituitary adenomas < 6–9 mm or in uncertain cases [ , ]. IPSS measures ACTH in the pituitary drainage versus peripheral venous sample; a central to peripheral ACTH gradient >2 before or >3 after stimulation (with CRH or desmopressin) is consistent with a pituitary source [ , ]. IPSS is highly sensitive and specific (∼95%) for differentiating pituitary from ectopic ACTH secretion, but is invasive and only available at specialized centers [ ]. Peripheral CRH and desmopressin tests are potential alternative noninvasive tests. They rely on robust ACTH and cortisol responses in CD versus a poor response in EAS. However, overlap in responses exists and the diagnostic accuracy of these tests is lower than IPSS [ ]. High dose dexamethasone suppression testing with 8 mg of dexamethasone has been used to differentiate between CD and EAS. In theory, patients with CD retain the ability to inhibit ACTH secretion in response to high dose dexamethasone, whereas ACTH production is not affected by dexamethasone in those with EAS [ ]. However, some patients with EAS (especially benign carcinoid tumors) do have ACTH suppression in response to high dose dexamethasone not making this test sufficiently accurate [ , ]. A combination of CRH and desmopressin testing, pituitary MRI, and whole-body CT has been proposed as an alternative to IPSS in specialized centers for selected patients [ ].
If the pituitary is confirmed to be the source of ACTH overproduction, then pituitary surgery is the first line treatment in most patients [ , ].
If an ectopic source is suspected, then localization imaging should be pursued. EAS is commonly caused by carcinoid tumors of the lung, islet cell tumors of the pancreas, medullary carcinoma of the thyroid, small cell tumors of the lung, and tumors of the thymus [ ]. CT scan of cervical, thoracic, abdominal, and pelvic regions is the initial imaging modality for localization of the tumor. In the cases of negative anatomic imaging and a strong suspicion for EAS, nuclear imaging ( 68 Ga DOTATATE- and/or FDG-PET) is recommended [ ].
A diagnostic algorithm for CS from the Pituitary Society Guidelines is depicted in Fig. 13.1 [ ].
Epidemiology of hypertension in Cushing’s syndrome
Elevated blood pressure is more prevalent in EAS, and one study reported it was more common in adrenal CS versus Cushing’s disease (CD) [ ]. The incidence is much lower in those with iatrogenic CS (20%), and in these patients, hypertension is usually dose dependent [ , ]. There is debate over or as to whether the severity of blood pressure elevation in CS is related to the magnitude of cortisol elevation [ , ]. Hypertension appears to be associated with older age and longer exposure to excess glucocorticoids [ ]. There appears to be no sex differences in hypertension prevalence among patients with CD [ ]. Most patients present with mild to moderate hypertension, but some patients (17%) have severe blood pressure elevations [ ]. On the other hand, the prevalence of CS is 0.5% in hypertensive patients and <1% in those with resistant hypertension [ ]. Thus, not all patients with hypertension should be screened for CS. Screening should occur in those with other clinical findings suggestive of a diagnosis of CS. Additionally many patients with CS may also have essential hypertension that predates the CS [ ].
The risk of cardiovascular disease is 4–5 fold higher in CS patients than in the general population, and risk factors include hypertension, hypercoagulability, obesity, dyslipidemia, and diabetes mellitus [ ]. Thus, proper evaluation and management of all other comborbidities is promptly needed after diagnosis.
Pathophysiology of hypertension in Cushing’s syndrome
Early in the disease process, there is an altered blood pressure circadian rhythm in patients with CS [ ], while circadian rhythm is preserved in those with essential hypertension or primary aldosteronism. The abnormal circadian rhythm in blood pressure elevation is seen in both untreated and treated patients, and often there is no nocturnal fall; some patients even have a rise in blood pressure at night. Interestingly, patients on exogenous glucocorticoids (GCs) also lose the diurnal variation [ ]. The absence of nocturnal blood pressure fall is associated with a higher rate of cardiovascular events, increased left ventricular hypertrophy, faster progression of microalbuminuria and renal damage, increased vascular stiffness, and an overall increased cardiovascular mortality [ ].
The pathogenesis of hypertension in CS is complex and not fully understood [ ]. There are several mechanisms for hypertension in those with CS including: MC receptor activation, activation of the renin-angiotensin system, increased sensitivity to vasoconstrictors, increased beta-adrenergic receptor sensitivity to catecholamines, suppression of vasodilators, sleep apnea, and insulin resistance [ , , ]. These mechanisms are summarized in Table 13.1 .
| Mineralocorticoid receptor activation | 11 β HSD2 saturation and decreased activity [ , , ] ↑ Plasma volume [ ] |
| Renin angiotensin system (RAS) activation | ↑ Angiotensinogen [ , ] ↑ Angiotensin II 1A receptors I in blood cell [ ] ↑ Pressor response to angiotensin II [ , , ] |
| Sympathetic nervous system | ↑ Sensitivity of β adrenergic receptors [ , ] |
| Vasoregulatory system | ↑ Endothelin 1 [ , , , ] (some studies indicate no role of ET1 [ , ]) ↓ Sodium calcium exchanger in vascular smooth muscle = ↑ cytosolic calcium = vasoconstriction [ , ] ↑ Erythropoietin [ ] ↓ Response to atrial natriuretic peptide [ ] ↓ Nitric oxide and L arginine [ , ] ↓ Prostacyclin [ ] ↓ Prostaglandin E2 [ ] ↓ Kallikrein [ ] ↑ VGEF [ ] |
| Other | Sleep apnea [ , ] Insulin resistance [ ] |
Cortisol binds to the glucocorticoid receptor (GR) and MC receptor. Activation of the MC receptor causes reabsorption of sodium in the kidneys, and intravascular volume expansion due to overactivity of the epithelial sodium channel (ENaC) [ , ]. Sodium reabsorption in the distal nephron is coupled with tubular secretion of potassium and protons, leading to hypokalemia and metabolic alkalosis [ ]. Hypokalemia leads to activation of the sodium chloride cotransporter (NCC) leading to further volume expansion [ ]. Volume expansion leads to lower levels of renin [ ]. The MC receptor has a similar binding affinity for both aldosterone and cortisol in vitro. However, in vivo 11-β-hydroxysteroid dehydrogenase-2 (11-βHSD2) converts cortisol to cortisone, which does not activate the MC receptor. This enzyme is overwhelmed in CS leading to impaired cortisol-to-cortisone metabolism. Of note, there is significant variability in the activity of 11-βHSD2, and there is evidence of a positive association between 11-βHSD2 activity and BMI [ ]. Additionally, in cultured human cells, ACTH decreased the activity of 11-βHSD2 in a dose-dependent manner [ , ]. Excess MC activity contributes directly to endothelial dysfunction, left ventricular hypertrophy, and cardiomyopathy [ ]. MC excess is most prevalent in patients with EAS and these patients often have hypokalemic alkalosis, which is only present in <10% of those with other etiologies of CS [ ]. Patients with EAS also have increased production of other adrenal metabolites such as deoxycorticosterone (DOC) and corticosterone, which cause sodium retention [ ]. Thus MC excess may not be a significant mechanism of hypertension in other forms of CS besides EAS [ ]. Supporting this is the fact that MC receptor antagonism does not fully normalize blood pressure in CS patients, suggesting that there are other mechanisms of hypertension [ ]. Long-term use of mifepristone, a GR blocker that does not bind the MC receptor, can normalize blood pressure in CS in some patients, while worsening it in others. Thus, it appears that activation of MC receptor and sodium retention can amplify hypertension but is not required for its development [ ].
Patients with CS have increased angiotensinogen levels , the renin substrate. However, plasma concentrations of angiotensin II, aldosterone, and renin are often normal and thus there is speculation of increased throughput in the renin-angiotensin-aldosterone (RAAS) system [ , ]. There are conflicting data on renin levels in CS patients with some studies showing normal and other studies showing low levels, while a few studies even show elevated levels. One study focused on patients with CD found low renin and aldosterone levels, thus suggesting, in these patients, hypertension is not renin-mediated [ ]. CS patients have an increased pressor response to angiotensin II; in rats, steroid exposure results in increased angiotensin II receptor type 1 expression in brain and peripheral tissues, and increases angiotensin II converting enzyme (ACE) [ ]. In humans with CS, there are greater number of angiotensin II 1A receptors in blood cells [ ]. Glucocorticoids also enhance the central actions of angiotensin II to increase blood pressure [ , ], and intravenous angiotensin II antagonist administration attenuates glucocorticoid induced hypertension [ ].
Glucocorticoids cause increased secretion of endothelin-1 (ET-1), a vasoconstrictor produced by smooth muscle cells and vascular endothelial cells [ ]. Levels are significantly elevated in patients with untreated CS [ ] and there is a decline in ET-1 in Cushing’s patients following remission [ ]. Studies of the role of ET-1 in rats with glucocorticoid induced hypertension have been conflicting. In animal studies, 11-βHSD2 inhibitor impairs cortisol-to-cortisone metabolism and increases vascular ET-1 levels. Chronic use of an endothelin type-A receptor antagonist normalizes vascular ET-1 concentration and reduces blood pressure, supporting a role of ET-1 in glucocorticoid mediated hypertension [ ]. However, bosentan (a nonselective endothelin antagonist) has no effect on blood pressure in rats with ACTH-induced hypertension, indicating there is no major role of ET-1 in Cushing’s syndrome-mediated hypertension [ , ].
Additionally, glucocorticoids downregulate the expression of the sodium-calcium exchanger in vascular smooth muscle [ ]. This results in higher cytosolic calcium levels causing vasoconstriction. High cortisol levels cause increased concentrations of erythropoietin (EPO) leading to polycythemia. EPO has a direct vasoconstrictor effect and may also contribute to elevated blood pressure [ ].
Cortisol results in the upregulation of the sympathetic nervous system by potentiating the action of catecholamines [ ]. Chromaffin cells convert norepinephrine to epinephrine using the enzyme phenylethanolamine N-methyltransferase (transcription is glucocorticoid dependent) [ ]. The sensitivity of β-adrenergic receptors is increased in states of cortisol excess, though catecholamine levels are normal. This results in increased vascular tone. Increased pressor response to angiotensin II and noradrenaline, and greater negative chronotropism to phenylephrine has been shown in patients with CS. Additionally, there is an increase in the number of α 1B -adrenergic receptors in vascular smooth muscle which further increases vascular tone [ ].
Glucocorticoids impair the action or production of vasodilators such as atrial natriuretic peptide (ANP), nitric oxide (NO), prostacyclin, prostaglandin E2 , and kallikrein [ ]. Circulating levels of ANP are normal or increased in CS; however, patients have a decreased response to ANP as compared with healthy subjects. Glucocorticoids inhibit the enzyme NO synthase leading to lower NO levels [ ]. Decreased NO activity is associated with increased production of vascular superoxide, which is important in the development of hypertension and atherosclerosis. There is also decreased transmembrane transport of L-arginine, the substrate for NO production. L-arginine has been shown to have beneficial endothelial effects [ ]. Glucocorticoids also inhibit production of prostacyclin [ ].
Plasma levels of vascular endothelial growth factor ( VGEF) , an important angiogenic factor, are increased in patients with CS. This leads to increased angiotensin converting enzyme activity in endothelial cells, and likely increased local angiotensin II production [ ].
With weight gain there is an initial reduction in urine sodium excretion . Additionally plasma volume, extracellular fluid volume, and exchangeable sodium are increased significantly and contribute to elevated blood pressure [ ].
Sleep apnea is also common in patients with CS. These patients have disproportionate weight gain from adiposity in the head, neck, and waist areas. There are also myopathic changes in genioglossal and geniohyoid muscles responsible for the patency of the airway which contributes to development of sleep apnea [ ]. Sympathetic tone is increased during hypoxemic episodes leading to hypertension. Regular use of continuous positive airway pressure (CPAP) has been found to markedly improve blood pressure in these patients [ ]. Central hypothyroidism is also common in patients with CS and can contribute to development of sleep apnea [ ].
Insulin resistance is also thought to play a role in the development of hypertension in CS patients [ ].
Other complications of Cushing’s syndrome
Other complications of CS include cardiovascular disease, hyperglycemia, hyperlipidemia, hypokalemia, hypercoagulability, infection, osteoporosis, infertility, and neuropsychiatric disorders [ , ].
Cardiovascular risk is markedly elevated in patients with CS. Dyslipidemia is present in 16%–64% and type 2 diabetes mellitus in 30%. The increased mortality in CS patients is driven primarily by myocardial infarction, stroke, and other vascular events [ ]. Predictors for mortality are older age at diagnosis, duration of active disease, and presence of diabetes and hypertension [ ]. This cardiovascular risk is reduced, but not normalized with treatment of CS [ ]. It is important to treat cardiovascular and metabolic complications simultaneously with treatment of CS itself in order to improve outcomes.
Patients with CS are at high risk for venous thromboembolic (VTE) and arterial thrombotic events which occur in about 18% [ ]. The incidence of venous thromboembolism is more than 10-fold increased as compared with the normal population [ , ]. The pathogenesis of hypercoagulability in CS is not completely understood; patients have increased production of procoagulant factors (factor VII and Von Willebrand factor ristocetin cofactor), and activation of the coagulation cascade [ ]. In patients who have had curative surgery for CD, the increased thromboembolic risk continues to persist in the first months after surgery. Thus, prophylactic anticoagulation has been recommended by many experts, particularly for patients with risk factors (history of VTE or thrombophilia, severe CS, estrogen/testosterone use, poor mobility, or prolonged hospitalization) [ , , , ]. The choice of regimen and duration is individualized; longer prophylaxis (up to 2 months post-operatively or longer) has been proposed for the highest risk patients and after bilateral adrenalectomy [ , ].
Patients with CS are also at risk of various bacterial, viral, and fungal infections due to suppression of the immune system. Pneumocystis jiroveci i pneumonia (PJP) is a high-mortality opportunistic fungal infection that sometimes develops in patients with severe CS. Elevated cortisol levels result in deprivation of T lymphocytes and macrophages [ ]. Additionally, treatment of CS leads to recovery of the T cells which can result in an inflammatory reaction to PJP, similar to immune reconstitution syndrome (IRIS) in HIV patients [ ]. The risk of PJP has been found to be associated with the degree of cortisol elevation. Prophylaxis with trimethoprim-sulfamethoxazole (TMP-SMX) should be considered in patients with severe CS [ ].
Patients with cortisol excess are at risk for osteoporosis and bone fractures. Osteopenia is present in 60%–80% and osteoporosis in 30%–65% of patients with CS [ ]. Fractures may be the first clinical manifestation of CS and occur most commonly at the thoracic and lumbar vertebrae [ , ]. Glucocorticoids inhibit osteoblast function, thereby suppressing bone formation [ ]. Glucocorticoids also inhibit calcium reabsorption in the kidney and reduce calcium absorption in the intestine, thus leading to secondary hyperparathyroidism, increased bone resorption and risk of kidney stones [ , ]. CS patients have suppressed sex and growth hormones, further contributing to decreased osteoblast function [ ]. Bone mineral density improves with treatment of CS, and may be completely reversed in some [ ]. Conventional osteoporosis treatment with calcium, vitamin D, and bisphosphonate may result in more rapid improvement in bone mineral density than treatment of hypercortisolism alone [ ].
Glucocorticoids can inhibit growth hormone secretion, thus many patients with CS have growth hormone deficiency (GHD) and low hepatic production of insulin-like growth factor 1 (IGF-1). In patients with CD, GHD is more prevalent in children, younger patients and/or women; notably, diabetes mellitus and hypertension are more prevalent in patients with both CS and GHD [ ]. A recent study found that low IGF-1 at 6 months was also predictive of more severe muscle weakness postremission of CS [ ]. GHD has been shown in a meta-analysis to increase mortality in patients with hypopituitarism of various causes [ ]. However, though GH replacement ameliorates several complications associated with metabolic syndrome, prospective trials failed to show reversal of metabolic syndrome or cardiovascular complications [ ].
Treatment of Cushing’s syndrome and associated hypertension
Once the source of cortisol or ACTH excess is identified, initial resection of the primary lesion is recommended. In the case of an adrenal adenoma causing CS, patients may undergo unilateral adrenalectomy. Virtually all patients are cured by adrenalectomy and mortality is low, especially with a laparoscopic approach [ , ]. Bilateral macronodular adrenal hyperplasia (BMAH) and primary pigmented adrenal nodular adrenocortical disease are treated with bilateral adrenalectomy (or unilateral adrenalectomy for BMAH in selected cases) [ ]. Patients with CD should undergo transsphenoidal surgery (TSS), unless they are not candidates due to high surgical risk, they decline surgery, or surgery is not possible. Remission rate varies in the literature, up to 80% for microadenomas, while remission for macroadenomas in specialized centers is up to 60% [ ]. Remission is more likely in patients who have a noninvasive microadenoma visible on preoperative imaging and histopathological analysis, a postoperative serum cortisol nadir <55 nmol/L (<2 μg/dL) and an experienced surgeon [ , ] . In EAS, the source of ACTH production should be actively sought and surgically resected if feasible [ , ]. For curative purposes, EAS tumors should be well localized on imaging and in an accessible area without distant metastases [ ]. It is hard to determine outcomes of surgery in patients with EAS given the rarity of EAS, different underlying etiology and varying degree of metastases. From one study, surgical cure is achieved in 83% of those with bronchial carcinoid [ ]; patients with squamous cell and thymic carcinoid causing EAS tend to have poor prognosis [ ].
The role of preoperative management of hypercortisolism in CS is unclear. A retrospective database study found no differences at 6-month follow-up in morbidity or remission rates between those treated with medical therapy prior to and those proceeded directly to surgery. However, patients with more severe clinical features were more likely to receive medical therapy, which confounds the analysis. Also, medical treatment is recommended in patients with occult EAS to treat acute and possibly potentially life-threatening morbidities caused be severe hypercortisolism [ ]. There is no specific guidance on preoperative blood pressure management for CS patients; noncardiac surgery guidelines recommend a preoperative blood pressure <180–200/100–110 mmHg [ ]. In the long-term, blood pressure goal is <130/80 mmHg for patients with increased cardiovascular risk (atherosclerotic cardiovascular disease, ASCVD >10%–15%) and diabetes, and <140/90 mmHg for patients without increased cardiovascular risk [ , ]. Additionally, management of hyperglycemia, hyperlipidemia, as well as antiplatelet therapy for secondary and, in some patients, for primary prevention of CVD is recommended to reduce potential cardiovascular events perioperatively and long term [ ]. Patients with multiple comorbidities due to CS are likely to benefit from multidisciplinary management in conjunction with endocrinology, primary care provider, surgery, and cardiology consultations in case of suspected CVD.
Medical therapy for CS is implemented when the surgery is not possible or is delayed, in patients with severe hypercortisolism while awaiting surgery, and in case of persistent CS after an unsuccessful surgery. Options for medical treatment include adrenal steroidogenesis inhibitors (Osilodrostat, Metyrapone, Levoketoconazole, Ketoconazole, Etomidate, Mitotane), pituitary directed drugs for CD (Pasireotide, Cabergoline) and GR blockers (Mifepristone). These medications and their effects on blood pressure are summarized in Table 13.2 . Radiotherapy is used for incompletely resected or significantly enlarging pituitary adenomas causing persistent CD [ , ]. Finally, bilateral adrenalectomy and subsequent replacement of adrenal steroids can be considered in cases where rapid eucortisolism is necessary, or when other therapies have failed, or are not appropriate [ , ].
| Medication | Mechanism of action | Doses | Effect on blood pressure | Possible side effects |
|---|---|---|---|---|
| Antihypertensives | ||||
| Captopril [ , ] Trandolapril | Angiotensin converting enzyme inhibition | Captopril 3.75 mg TID Trandolapril 2 mg/day | Trandolapril ↓ in 50% Captopril and ramipril ↓ | Dry cough, hyperkalemia, angioedema |
| Losartan [ , ] Saralasin a | Angiotensin receptor blockers | Losartan 50 mg | Losartan ↓ in 50% Saralasin ↓ | Hyperkalemia, angioedema |
| Spironolactone [ , , ] | Mineralocorticoid receptor antagonist Androgen receptor blocker | 25–100 mg/day | May not affect blood pressure significantly | Hyperkalemia Irregular or absent menses Gynecomastia, reduced libido, erectile dysfunction |
| Eplerenone [ , ] | Mineralocorticoid receptor antagonist | 50–200 mg/day | No studies in CS | Hypokalemia Fewer androgen side effects than spironolactone |
| Triamterene [ ] | Potassium sparing diuretic Blocks epithelial sodium channel | 50–100 mg/day | No studies in CS | Dizziness, fatigue, hypokalemia, diarrhea |
| Chlorthalidone Hydrochlorothiazide (HCTZ) [ , , ] | Diuretic Impairs sodium transport in distal convoluted tubule | HCTZ 6.25–50 mg/day | No studies in CS | Hypokalemia, hyperuricemia, diabetes May reduce kidney stones in CS patients |
| Isosorbide Mononitrate [ ] | Donor of nitric oxide | 60 mg | ↓ in patients given GC | Headache, tachycardia, flushing, nausea |
| Atenolol [ , , ] Carvedilol Labetalol Nebivolol | Selective beta blockers [ ] | Carvedilol 25 mg BID Nebivolol 2.5–10 mg/day | No change in patients given GC Consider in patients with hx of MI, active angina, HF | Bradycardia, fatigue Avoid in patients with pheochromocytoma |
| Doxazosin [ , ] | Alpha 1 receptor antagonist causing vasodilation | 1–16 mg/day (usual dose 7 mg) | No specific studies in CS 3rd line therapy in resistant HTN | Dizziness, fatigue, headache, vertigo, edema |
| Medications used for Cushing syndrome treatment [ ] | ||||
| Pituitary directed drugs | ||||
| Cabergoline [ ] | D2 receptor agonist | 0.5–7 mg/week | Relaxes vascular smooth muscle ↓ cortisol in some patients ↓BP in some patients | Dizziness, nausea, mood changes, valvular heart disease (high dose) |
| Bromocriptine [ ] Not used for CS anymore, rarely used for Nelson’s syndrome | D2 receptor agonist | 2.5–40 mg/day | Variable reports on change in cortisol ↓ BP in non-CS patients | Nasal congestion, nausea, and postural hypotension |
| Pasireotide [ , , ] | Somatostatin receptor ligand (SSTR5 especially) | 600–900 µg BID 10–30 mg monthly (long acting) | ↓ BP even with partial UFC control | Nausea, gall stones, diarrhea, prolonged QT c , hyperglycemia (frequent) |
| Retinoic Acid [ , , , ] | ↓ ACTH production through inhibition of AP-I and Nur77/Nurrl transcriptional activities | 10–80 mg/day | ↓ cortisol ↓ BP in some patients | Conjunctival irritation, cheilitis, mucositis, nausea, headache, arthralgias, elevated triglyceride, transaminitis |
| Steroidogenesis inhibitors | ||||
| Osilodrostat [ , ] | Inhibits CYP11B1 and aldosterone synthase | 1–30 mg BID (doses required for most patients are 2–14 mg/day) | ↓ cortisol and aldosterone but ↑ DOC BP ↓ [ , ] but no change in one study [ ] | Hypokalemia, edema, HTN, hirsutism, nausea, diarrhea, adrenal insufficiency, QT prolongation |
| Metyrapone [ , , ] | Inhibitor of 11 hydroxylase and aldosterone synthase Inhibits CYP11B1 mainly), CYP17, CYP11B2, CYP19 | 0.5–4.5 g (3–4 x/day) | ↓ cortisol ↑ DOC BP: ↓ [ ], ↑ [ ] or no change [ ] | Hypokalemia, HTN, edema, hirsutism, alopecia, acne, clitoromegaly, voice deepening, adrenal insufficiency, gastrointestinal distress |
| Ketoconazole [ , ] | Inhibits StAR b , CYP11A1, CYP11B1, CYP17 | 400–1600 mg (BID or TID administration) | BP control appears to be related to cortisol control BP ↓ in 40% of patients or more | Adrenal insufficiency, transaminitis, gastrointestinal distress, hypogonadism, drug–drug interactions |
| Levoketoconazole [ ] | Inhibits CYP17A1, CYP11A1, CYP11B1, and CYP21A2 | 150–600 mg BID | No change in BP overall, though BP would improve in some patients with UFC decreased | Nausea, headache, adrenal insufficiency, transaminitis, gastrointestinal distress, drug–drug interactions, QT prolongation |
| Etomidate [ , , ] | Inhibits StAR b , CYP11A1, CYP11B1, CYP17 | Bolus 5 mg (1 x) followed by IV 0.02–0.3 mg/kg/hr | ↓ BP | Sedation, adrenal insufficiency, propylene glycol toxicity |
| Mitotane [ , , ] | Inhibits StAR b , CYP11A1, CYP11B1, CYP11B2, 3β-HSD Adrenolytic | 2–5 g/day | ↓ cortisol ↓ aldosterone No change in BP in one study | Adrenal insufficiency, dizziness, altered cognition, gastrointestinal distress, teratogenic, cytopenia, hyperglycemia low T4 m, drug–drug interactions |
| Glucocorticoid receptor antagonist | ||||
| Mifepristone [ , , ] | Glucocorticoid receptor antagonist | 300–1200 mg/day | ↓ action of cortisol on GR ↑ action of cortisol on MR ↓BP in some, ↑ in some and treat with MR blocker | Hypokalemia, HTN, adrenal insufficiency, vaginal bleeding, endometrial hyperplasia, nausea, fatigue, edema, arthralgias, edema, QTc prolongation |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


