Thrombocytopenia, usually defined as a platelet count of less than 150,000/μL, is a common reason for a hematology consult in both the inpatient and outpatient setting. In most patients, the cause of the thrombocytopenia can be identified and treated. This article reviews the clinical approach to the patient with thrombocytopenia, the mechanisms that underlie it, and the laboratory tests available to investigate it. A practical approach to the investigation and management of thrombocytopenia in the clinical settings commonly encountered by the hematology consultant is then described.
Thrombocytopenia, usually defined as a platelet count of less than 150,000/μL, is a common reason for a hematology consult in both the inpatient and outpatient setting. In most patients, the cause of the thrombocytopenia can be identified and treated. This article reviews the clinical approach to the patient with thrombocytopenia, the mechanisms that underlie it, and the laboratory tests available to investigate it. A practical approach to the investigation and management of thrombocytopenia in the clinical settings commonly encountered by the hematology consultant is then described.
Pertinent history in the patient with thrombocytopenia
Thrombocytopenia classically causes mucosal-type bleeding, and patients should be asked about epistaxis, gingival bleeding, and menorrhagia in women. Other bleeding manifestations may include petechiae, bruising, hematochezia, and melenic stools. A history of bleeding associated with past hemostatic challenges must be obtained, including surgeries, dental procedures, trauma, and child birth. The patient’s past and present alcohol use should be documented, and the patient should be asked about a history of liver disease, cirrhosis, jaundice, and risk factors for human immunodeficiency virus (HIV) and hepatitis infections. The medication history must also include questions about over-the-counter medications, herbal supplements, and the consumption of tonic water, which contains quinine, a cause of immune thrombocytopenia. Specific questions about the presence of a family history of bleeding are important, as congenital thrombocytopenia and Von Willebrand disease (VWD) can be diagnosed in young adults (reviewed in Ref. ) ( Table 1 ).
| History |
|
| Physical examination |
|
Physical examination
The physical examination in a patient with thrombocytopenia must establish first and foremost if there is evidence of bleeding associated with the thrombocytopenia, and whether the bleeding is in proportion to the degree of thrombocytopenia. Patients with platelet counts greater than 50,000/μL do not exhibit spontaneous bleeding unless a coagulopathy is also present, the platelets are dysfunctional, and/or the patient is on a platelet inhibitor. The first sign of spontaneous bleeding from thrombocytopenia is usually petechiae on dependent areas such as the lower legs, or in areas of pressure (eg, at the site of a blood pressure cuff). Other signs of bleeding associated with more severe thrombocytopenia include ecchymosis and blood-filled blisters on the oral mucosa. The physical examination may also establish the underlying condition causing the thrombocytopenia, such as enlarged lymph nodes and spleen in lymphoma, and splenomegaly, telangiectasias, palmar erythema, jaundice, and other stigmata of chronic liver disease in patients with cirrhosis (reviewed in Ref. ; see Table 1 ).
Physical examination
The physical examination in a patient with thrombocytopenia must establish first and foremost if there is evidence of bleeding associated with the thrombocytopenia, and whether the bleeding is in proportion to the degree of thrombocytopenia. Patients with platelet counts greater than 50,000/μL do not exhibit spontaneous bleeding unless a coagulopathy is also present, the platelets are dysfunctional, and/or the patient is on a platelet inhibitor. The first sign of spontaneous bleeding from thrombocytopenia is usually petechiae on dependent areas such as the lower legs, or in areas of pressure (eg, at the site of a blood pressure cuff). Other signs of bleeding associated with more severe thrombocytopenia include ecchymosis and blood-filled blisters on the oral mucosa. The physical examination may also establish the underlying condition causing the thrombocytopenia, such as enlarged lymph nodes and spleen in lymphoma, and splenomegaly, telangiectasias, palmar erythema, jaundice, and other stigmata of chronic liver disease in patients with cirrhosis (reviewed in Ref. ; see Table 1 ).
Ancillary tests
A complete blood count with a white blood cell differential is the first step in the workup of thrombocytopenia, and establishes whether the patient has other cytopenias and/or abnormal circulating cells. A review of the peripheral blood smear is essential, as it will rule out pseudothrombocytopenia (see later discussion), and enable the evaluation of all 3 cell lines for morphologic abnormalities. More specialized tests are ordered based on the initial clinical and laboratory evaluations, and are listed in Table 2 . Platelet-associated antibodies and reticulated platelet counts, although widely available, lack standardization and, in the authors’ opinion, are rarely helpful in establishing the cause of the thrombocytopenia.
| Test | Use |
|---|---|
| Complete blood count |
|
| Peripheral blood smear |
|
| PT and PTT Fibrinogen, D-dimer |
|
| Reticulated platelet count and antiplatelet antibodies |
|
| Heparin-associated antiplatelet factor 4 and serotonin release assay |
|
| HIV and hepatitis C serology |
|
| ANA, anti–double-stranded DNA |
|
| Anticardiolipin antibody, lupus anticoagulant, anti-β2 glycoprotein 1 |
|
| Drug-associated increase in antiplatelet IgG |
|
| Flow cytometry of peripheral blood |
|
| Serum protein electrophoresis |
|
| Bone marrow aspirate and biopsy |
|
| Ultrasonogram of liver and spleen |
|
| Liver/spleen scan |
|
Mechanisms of thrombocytopenia
Platelets are produced from proplatelets, which are long, branching processes that extend from the cytoplasm of mature megakaryocytes. Their production is regulated by cytokines, especially thrombopoietin (TPO), and by close interactions with the bone marrow stroma. Approximately 4 to 7 days are required for the megakaryocyte progenitor cell to mature, at which point it produces 1000 to 3000 platelets before its residual nuclear material is engulfed by macrophages. Platelets normally circulate for approximately 10 days, although this life span is decreased to 5 to 7 days in patients with thrombocytopenia. Disruption of any part of this process, when severe enough, can lead to thrombocytopenia.
When evaluating a patient with thrombocytopenia, the first step is to rule out pseudothrombocytopenia, a laboratory artifact with no clinical significance. True thrombocytopenia can be secondary to increased destruction of platelets, decreased production, sequestration (usually in the spleen), and hemodilution ( Table 3 ). Although in most clinical situations a predominant mechanism can be identified, in many patients more than one pathway contributes to the thrombocytopenia.
| Mechanism | Examples |
|---|---|
| Increased destruction (immune mediated) |
|
| Increased destruction (non–immune mediated) |
|
| Decreased production |
|
| Sequestration |
|
| Dilutional |
|
Pseudothrombocytopenia
Pseudothrombocytopenia, also called spurious thrombocytopenia, is an ex vivo phenomenon occurring in 0.09% to 0.29% of the population, in which platelets clump when blood is anticoagulated with a calcium chelator such as ethylenediaminetetraacetic acid (EDTA). Automated counters do not correctly identify the clumps as platelets, and as a result the platelet count is reported as low. Furthermore, the platelet clumps are sometimes misrecognized as neutrophils, causing pseudoleukocytosis. Review of a peripheral blood smear ( Fig. 1 ) demonstrates the clumps, and the automated platelet count is usually higher when blood is collected into an alternative anticoagulant such as citrate or heparin. Pseudothrombocytopenia is caused by circulating antiplatelet antibodies against platelet membrane glycoproteins that are modified by the exposure to anticoagulants. Platelet satellitism is a less common form of pseudothrombocytopenia in which EDTA causes antibodies against glycoprotein (GP) IIb/IIIa to attach platelets to neutrophils and monocytes via the leukocyte Fcγ receptor III. The peripheral blood smear ( Fig. 2 ) demonstrates platelets forming rosettes around the neutrophils and/or monocytes.
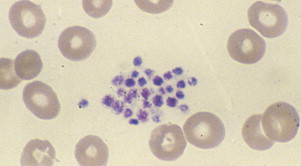
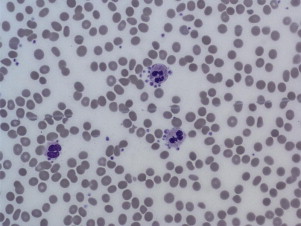
There are no known pathologic effects of the presence of the antiplatelet antibodies associated with pseudothrombocytopenia, and it is important to establish and document the diagnosis so that patients are not subjected to unnecessary testing and treatment.
Thrombocytopenia from Increased Platelet Destruction
The normal platelet life span of 7 to 10 days can be shortened by both immune-mediated and non–immune-mediated processes, and, when the destructive process overwhelms the bone marrow’s ability to increase platelet production, the result will be thrombocytopenia.
Immune Destruction of Platelets
In patients with immune-mediated thrombocytopenia, antibodies attach to platelets and promote their destruction by the reticuloendothelial system. These antibodies can be either autoantibodies or alloantibodies. The most common cause of autoimmune platelet destruction is immune thrombocytopenic purpura (ITP), which is usually caused by antibodies of the immunoglobulin G subtype directed against platelet membrane glycoproteins. In heparin-induced immune platelet destruction, autoantibodies target a heparin-platelet factor 4 complex and cause thrombocytopenia by binding to the membrane Fc receptor and activating the platelets (see Table 3 ).
Alloimmune destruction is a rare but severe cause of thrombocytopenia, in which platelets are destroyed by alloantibodies usually targeting the platelet antigen HPA-1a (PlA1) that have been transmitted through a transfusion (see discussion of posttransfusion purpura).
Nonimmune Destruction of Platelets
Nonimmune destruction of platelets occurs in patients with thrombotic microangiopathies such as thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, and the elevated liver enzymes, low-platelet (HELLP) syndrome of pregnancy. In these patients thrombocytopenia results from the shearing of platelets in a damaged microvasculature filled with thrombi. Microangiopathic hemolytic anemia is also present, and coagulation parameters are normal or near normal (see Table 3 ).
Disseminated intravascular coagulation (DIC) is a common cause of thrombocytopenia in acutely ill patients, and here the platelet destruction is secondary to their activation by thrombin and proinflammatory cytokines (see Table 3 ). Patients who have undergone cardiopulmonary bypass, insertion of an intra-aortic balloon pump, or insertion of a ventricular assist device develop thrombocytopenia as a result of the activation and destruction of platelets exposed to the artificial surfaces (see later discussion). Patients with a severely abnormal endovascular surface such as a damaged, malfunctioning heart valve, aortic aneurysm, or a vascular malformation may also develop thrombocytopenia secondary to activation of platelets on the abnormal surface.
Decreased Production of Platelets
Multiple processes affecting megakaryocyte maturation and differentiation, either directly or through a more global effect on hematopoiesis, may result in thrombocytopenia, often accompanied by anemia and/or leukopenia (see Table 3 ).
Sequestration of Platelets
Hypersplenism is defined as anemia, leukopenia, and/or thrombocytopenia that are caused by abnormal splenic-mediated destruction often associated with splenomegaly. Normally, approximately 30% of the platelet mass resides in the spleen. This percentage increases with increased spleen size, and most patients with splenomegaly will have thrombocytopenia. When the cause of the thrombocytopenia is hypersplenism, the platelet count will rarely drop below 20,000/μL, and the low platelet count is not usually associated with bleeding unless other hemostatic abnormalities are present (see also Table 1 ). Thrombocytopenia is usually the first manifestation of hypersplenism in patients with liver disease, and the hematologist sometimes establishes the diagnosis of otherwise clinically occult cirrhosis during the workup of a low platelet count.
Dilutional Thrombocytopenia
Thrombocytopenia develops in patients receiving infusions of large amounts of non–platelet-containing fluids. A rapid transfusion of 10 to 12 units of packed red blood cells will result in a platelet drop of approximately 50%.
Thrombocytopenia in the internal medicine patient
This section focuses on the different causes of thrombocytopenia that the clinician may encounter in either the outpatient or inpatient medical setting.
Nutritional Deficiencies
Vitamin B 12 and folate deficiency cause megaloblastosis and pancytopenia through inhibition of purine synthesis. Thrombocytopenia may be the predominant cytopenia, and can be severe (reviewed in Ref. ). Because these are common, readily treatable conditions, all patients with thrombocytopenia should have documentation of normal levels of vitamin B 12 and folic acid as part of their workup. Repletion of the deficient vitamin will rapidly correct the thrombocytopenia, and it is the authors’ practice to offer repletion to all patients with low or borderline levels. Folate deficiency is commonly associated with ethanol abuse, and the etiology of thrombocytopenia may be multifactorial in these patients (see later discussion).
Iron Deficiency
Iron deficiency is usually associated with thrombocytosis. In rare cases, however, thrombocytopenia may be present, and in these patients both increased and decreased numbers of megakaryocytes in the bone marrow have been reported. Although the exact mechanism remains unclear, the observation of increased megakaryocytes in the marrow accompanied by a rapid increase in platelets after iron administration suggests the involvement of iron in late-stage thrombopoiesis.
Alcohol Abuse and Thrombocytopenia
Thrombocytopenia is the most common hematologic abnormality in patients who abuse alcohol. Alcohol ingestion may lead to acute thrombocytopenia, and a common scenario is a falling platelet count in a patient admitted after binge drinking. With alcohol cessation and adequate folate and vitamin B 12 repletion, recovery of platelet counts occurs within 1 to 2 weeks and may even be associated with a rebound thrombocytosis.
Heavy alcohol consumption has direct toxic effects on the bone marrow and causes a reversible suppression of platelet production. Peripheral blood smear examination reveals thrombocytopenia, and may also show macrocytosis and stomatocytes. Marrow examination usually reveals a normal megakaryocyte count, but their number can also be significantly reduced. Because alcohol inhibits heme biosynthesis, ringed sideroblasts are common.
Liver Disease and Thrombocytopenia
Thrombocytopenia in patients with liver disease is multifactorial. Chronic thrombocytopenia in a patient with liver cirrhosis is usually a result of portal hypertension and splenomegaly, leading to splenic sequestration. Splenomegaly can be associated with any combination of cytopenia(s), including isolated thrombocytopenia. There may also be direct marrow-suppressive effects from the cause of the liver damage, whether from alcoholism or viral or immune hepatitis. Hepatitis C and autoimmune hepatitis are associated with immune-mediated thrombocytopenia. Chronic liver disease can also lead to deficiency of thrombopoietin, the hematopoietic growth factor responsible for platelet production. In addition, DIC leading to platelet consumption is seen in severe liver disease. Indeed, the combined hematologic effects of ongoing alcohol abuse in patients with cirrhosis who have pancytopenia and coagulopathy are particularly severe, with bleeding complications often necessitating significant transfusion support.
The Congenital Thrombocytopenias
Although congenital thrombocytopenias are rare, this is an essential diagnosis to establish, because patients are often misdiagnosed as having ITP, and subjected to unnecessary and potentially harmful treatments. Some important considerations for diagnosing congenital thrombocytopenias are the age of onset and chronicity of symptoms. Patients with inherited conditions associated with severe thrombocytopenia are generally recognized shortly after birth. Patients with milder forms of thrombocytopenia may present later in life, particularly during times of hemostatic challenge (eg, onset of menses, childbirth, trauma, surgery). Many other patients with mild to moderate thrombocytopenia, however, may be clinically asymptomatic and present only after routine blood tests.
If congenital thrombocytopenia is suspected, further evaluation is guided by platelet size. When a male patient presents with small platelets, an X-linked thrombocytopenia such as Wiskott-Aldrich syndrome should be considered. For patients presenting with normal-sized platelets, the differential includes neonatal conditions such as congenital amegakaryocytic thrombocytopenia, thrombocytopenia with absent radii, and a familial platelet disorder with predisposition to acute myeloid leukemia. Large platelets are seen in several inherited disorders of platelets including VWD 2B, MYH9-related diseases such as the May-Hegglin anomaly, Bernard-Soulier syndrome, and the gray platelet syndrome.
Von Willebrand Disease 2B
VWD 2B, also known as platelet-type VWD, deserves special mention as patients may present for the first time in adulthood. This rare autosomal dominant disorder is characterized by the presence of ultralarge multimers of Von Willebrand factor (VWF), which bind preferentially to platelets, leading to clumping and mild, fluctuating thrombocytopenia. VWD 2B, like other forms of VWD, presents clinically with excessive mucous membrane, menstrual, and/or postpartum hemorrhage.
Review of a peripheral blood smear may show pseudothrombocytopenia (see earlier discussion), and the platelet size is normal or large. Laboratory tests show a disproportionate reduction in VWF activity assays relative to VWF antigen, and absence of high molecular weight multimers. Desmopressin should be avoided, as it may lead to worsening thrombocytopenia caused by the increase in VWF multimers and increased platelet agglutination and clearance.
MYH9-related diseases
The MYH9-related diseases are associated with macrothrombocytopenia and involve myosin IIA mutations. The May-Hegglin anomaly is the most common form. The peripheral smear shows giant platelets and Döhle-like bodies in neutrophils ( Fig. 3 ). Associated features include cataracts, hearing loss, and renal failure. Automated counters often underestimate the true platelet count because of limitations from preset size restrictions. Although the platelet count is variable and sometimes less than 20,000/μL, platelet function is normal and patients are usually asymptomatic.
Bernard-Soulier syndrome
Bernard-Soulier syndrome, caused by an absent GP Ib-V-IX complex on the platelet surface, leads to bleeding out of proportion to the platelet count. Similar to the MYH9-related diseases, the peripheral smear demonstrates giant platelets. In homozygous patients the diagnosis is made by platelet aggregation studies showing lack of aggregation to ristocetin and a negative VWD workup. Flow cytometry to quantify platelet glycoproteins is particularly helpful in identifying heterozygote patients.
Drug-induced thrombocytopenia
Drug-induced thrombocytopenia (DITP) is an important entity to consider in all patients presenting with thrombocytopenia, and included in this discussion is thrombocytopenia caused by over-the-counter supplements and food items. The onset may be sudden and severe (platelet count nadirs are often <20,000/μL), leading to significant morbidity and even mortality if the offending agent is not promptly removed. DITP can be misdiagnosed as idiopathic ITP, and the underlying etiology may be missed if patients are not specifically asked about foods, beverages, and/or herbal supplements.
DITP can be caused by nonimmune (eg, valproic acid or chemotherapy resulting in myelosuppression) or, more commonly, immune-mediated processes. The immunologic mechanisms include autoantibody production (eg, gold [Ridaura; Solganal, others]), drug glycoprotein complex(es) (eg, quinine [Quinamm; Quindan, others]), ligand-induced binding (eg, eptifibatide [Integrilin]), and hapten-dependent binding (eg, penicillin) (reviewed in Ref. ). The mechanism for heparin-induced thrombocytopenia (HIT) is unique, as antibodies against platelet factor 4–heparin complexes cause platelet activation, thrombocytopenia, and thrombosis, rather than bleeding (see Table 3 ).
Drug-dependent antiplatelet antibodies usually occur within 1 to 2 weeks of exposure to the offending drug, but in some instances may develop after chronic, intermittent exposure (reviewed in Refs. ). On rare occasions acute immune-mediated thrombocytopenia may develop within several hours of exposure, as in the case of GP IIb/IIIa inhibitors used in cardiac procedures (see the section on thrombocytopenia in the cardiac care unit). Regardless of the specific mechanism, DITP occurs only in the presence of the offending agent, and removal of the drug should lead to improvement within 1 to 2 days, with resolution of the platelet count within 1 to 2 weeks. Indefinite avoidance of the offending agent is recommended. If ITP was initially suspected and corticosteroids were initiated, therapy may be stopped once the offending agent has been removed and the platelet count normalizes.
DITP should be suspected when a patient presents with severe thrombocytopenia of unclear etiology, particularly in patients with repeated episodes and prompt resolution. In a cohort of 343 patients diagnosed as ITP, 8% were subsequently diagnosed with DITP, with quinine (including tonic water) being the most common cause. The 2 most common causes of thrombocytopenia in one case-controlled study were trimethoprim/sulfamethoxazole (Bactrim; Septra) and quinine, in 38 and 26 cases/10 6 users/week, respectively.
A systematic review of all published reports of DITP as of October 2010 is available online at www.ouhsc.edu/platelets . A causal relationship between a drug and thrombocytopenia is defined in that database if there is a single report with definite evidence or at least 2 reports with probable evidence. A modified list of the most common drugs causing thrombocytopenia is also provided in Table 4 . There is currently no published comprehensive list of foods or herbal remedies associated with thrombocytopenia, but convincing case reports suggest that tahini (pulped sesame seeds), Lupinus termis beans, Jui Chinese herbal tea, cow’s milk, and cranberry juice can cause thrombocytopenia.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


