Abstract
Inherited disorders in the metabolism and recognition of vitamin D cause hypocalcemia, hypophosphatemia, and severe skeletal deformities classic for rickets. Mutations of the 25-hydroxyvitamin d -1-hydroxylase are responsible for pseudovitamin D resistant rickets. Mutations of the vitamin D receptor are responsible for vitamin D deficiency rickets. Alterations in phosphate metabolism can also impair the 25-hydroxyvitamin d -1-hydroxylase (CYP27B1). Patients with X-linked hypophosphatemic rickets and autosomal dominant hypophosphatemic rickets have an alteration in the metabolism of fibroblast growth factor 23 and possibly other phosphatonins, leading to severe phosphate wasting in the kidneys and reduced production of 1,25-dihydroxyvitamin D. A mutation of the 25-hydroxyvitamin d -24-hydroxylase (CYP24A1) is a cause for idiopathic infantile hypercalcemia, due to an unregulated increase in intestinal calcium absorption due to the lack of breakdown of 1,25-dihydroxyvitamin D. Various treatment strategies are described to help mitigate the calcium and phosphate metabolism disorders and the skeletal consequences of these inherited disorders.
Keywords
vitamin D, vitamin D resistance, rickets, osteomalacia, vitamin D-dependent rickets, hypophosphatemic rickets, 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, 1-hydroxylase, 24-hydroxylase, idiopathic infantile hypercalcemia, fibroblast growth factor 23
Introduction
Vitamin D is essential for bone health. It is responsible for maintaining calcium and phosphorus metabolism that results in the development and maintenance of a healthy skeleton. Genetic defects in the metabolism and recognition of vitamin D lead to alterations in calcium, phosphorus, and bone metabolism. The severity of the bone disease associated with these genetic defects depends on the severity of the consequences of the genetic defect on calcium and phosphorus metabolism. In children there is often poor mineralization of the bone leading to classic rachitic changes in the skeleton. The skeletal deformities often process throughout adulthood. The goal of this chapter is to provide a perspective on the causes and consequences of these genetic defects on calcium, phosphorus, and bone metabolism and to provide treatment strategies.
Calcium, phosphorus, and vitamin D metabolism
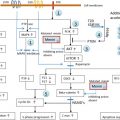
Vitamin D is recognized as the sunshine vitamin because it is made in the skin during exposure to sunlight. During exposure to sunlight, the ultraviolet B photons are absorbed by 7-dehydrocholesterol in the epidermis and dermis, resulting in the production of previtamin D 3 ( Fig. 13.1 ). Once formed in the plasma membrane of the skin cell it rapidly isomerizes to vitamin D 3 . Vitamin D 3 is ejected out of the plasma membrane into the extracellular space, whereby diffusion enters the derma capillary bed for transport on the vitamin D binding protein (DBP) to the liver. Vitamin D coming from the diet is incorporated into the chylomicrons and absorbed into the lymphatic system, which then enters into the venous blood supply. There are two forms of vitamin D. Vitamin D 2 comes from the UVB irradiation of yeast. Vitamin D 3 is made from 7-dehydrocholesterol obtained from sheep’s lanolin. Physiologic doses of vitamin D 2 and vitamin D 3 are equally effective in maintaining vitamin D status (D represents either D 2 or D 3 ).
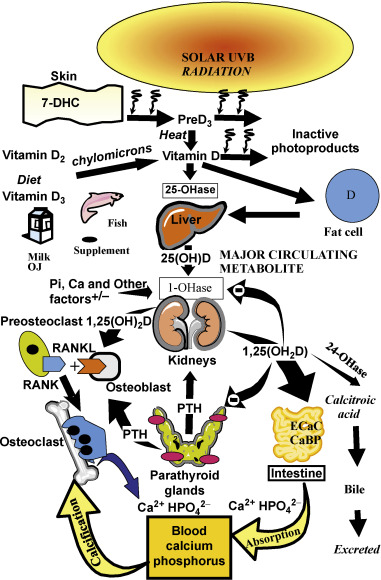
Vitamin D is biologically inert and requires a hydroxylation on carbon 25 by vitamin D-25-hydroxylase to form the major circulating form of vitamin D 25-hydroxyvitamin D (25(OH)D) ( Fig. 13.1 ). This metabolite is also biologically inert and requires further activation in the kidneys on carbon 1 by the 25-hydroxyvitamin d -1-hydroxylase (cyp27B1;1-OHase) to form 1,25-dihydroxyvitamin D (1,25(OH) 2 D). Vitamin D 2 and vitamin D 3 are metabolized in the liver and kidneys in a similar manner.
1,25(OH) 2 D interacts with its nuclear receptor, the vitamin D receptor (VDR), in the small intestine to increase the efficiency of intestinal calcium absorption. In a vitamin D deficient state, the small intestine is able to absorb passively about 10–15% of dietary calcium. Vitamin D sufficiency enhances the absorption of calcium to 30–40%. 1,25(OH) 2 D also plays an important role in phosphorus metabolism by enhancing the efficiency of phosphorus absorption in the jejunum and ilium. The small intestine passively absorbs about 60% of dietary phosphate, and vitamin D sufficiency increases it to about 80%. When there is adequate calcium and phosphorus in the diet, vitamin D is able to maintain the serum calcium in a normal range of 8.6–10.2 mg/dL and the serum phosphorus level at 2.5–4.5 mg/dL. 1,25(OH) 2 D interacts with its receptor in the kidneys to enhance calcium and phosphate reabsorption. It is the calcium phosphate product in the circulation and in the extravascular space that is critically important for the mineralization of osteoid laid down by osteoblasts.
When dietary calcium is inadequate to maintain extracellular calcium concentrations, 1,25(OH) 2 D interacts with its VDR in osteoblasts to increase the expression of receptor activator of NFκB ligand (RANKL). This sets into motion the ability of the osteoclast to dock with the RANK receptor on the preosteoclast, resulting in the formation of a multinucleated mature osteoclast ( Fig. 13.2 ). The mature osteoclast releases HCl and collagenases resulting in the destruction of the mineral and matrix releasing calcium and phosphorus into the circulation.
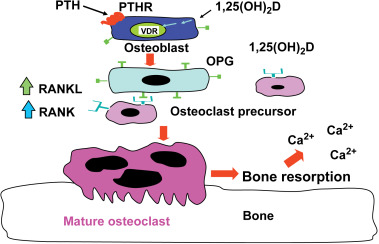
The interaction of 1,25(OH) 2 D with its VDR in osteoblasts also results in the expression of osteocalcin and alkaline phosphatase, which play an important role in bone metabolism. However, 1,25(OH) 2 D does not appear to play a direct role in the mineralization process. It is known in both rodents and humans that vitamin D is not necessary for the mineralization of osteoid matrix. This was demonstrated when vitamin D deficient rats were either infused with calcium and phosphorus to maintain a normal calcium–phosphorus product in the circulation or when they received a high calcium lactose, high phosphorus diet that maintained a normal serum calcium–phosphorus product. In both studies, bone development and mineralization occurred in a normal manner similar to animals that were vitamin D sufficient.
Vitamin D deficiency and rickets
Vitamin D deficiency is the most common cause of rickets and osteomalacia. Because vitamin D plays such an important role in maintaining serum calcium, it was assumed that the mechanism by which vitamin D deficiency causes a mineralization defect of the osteoid is due to hypocalcemia. However, the body maintains its serum calcium in the normal range in order to maintain a wide variety of metabolic functions and neuromuscular activity, and thus most patients who are vitamin D deficient have a normal serum calcium due to the increased bone calcium mobilization by 1,25(OH) 2 D and parathyroid hormone (PTH) ( Figs 13.1 and 13.2 ). In a vitamin D deficient state, ionized calcium declines, causing an immediate stimulus to the calcium sensor in the parathyroid glands, resulting in an increase in the expression and production of PTH. PTH increases tubular reabsorption of calcium in the kidneys and increases the expression of RANKL in osteoblasts, resulting in an increased number of osteoclasts, which remove precious calcium stores from the skeleton and increases the kidneys’ production of 1,25(OH) 2 D. PTH also causes the internalization of the sodium-dependent phosphate cotransporter (NaPi-2A), resulting in loss of phosphate into the urine. This results in a lowering of the serum phosphorus level, causing an inadequate calcium–phosphorus product needed for matrix mineralization. Thus, the major cause of osteomalacia and rickets is due to secondary hyperparathyroidism and low–normal or low serum phosphorus level.
Genetic causes of rickets – osteomalacia: disorders in vitamin D metabolism and recognition
Vitamin D-25 Hydroxylase Deficiency
There are at least four different hepatic enzymes found in the mitochondria and microsomes that are capable of metabolizing vitamin D to 25(OH)D. This is the likely reason why there have only been rare reports of a 25-hydroxylase deficiency causing rickets in children.
Vitamin D-Dependent Rickets Type I: Pseudovitamin D Deficiency Rickets (PDDR)
Before the discovery that vitamin D needed to be metabolized in the liver and kidneys before it could carry out its biologic actions on calcium and phosphorus metabolism, it had been reported that there were children who had rickets and who did not respond to physiologic doses of vitamin D. The children had hypocalcemia, hypophosphatemia, elevated alkaline phosphatase, elevated PTH, and severe rachitic changes on X-ray. However, many of these children responded to very large pharmacologic doses of vitamin D. As a result, these children received the diagnosis of vitamin D-dependent rickets because of their need for much higher amounts of vitamin D to treat their rickets. With the revelation that vitamin D needed to be metabolized in the kidneys to its active form, it was speculated that vitamin D-dependent rickets was caused by a mutation of the 1-OHase, resulting in either inadequate production of 1,25(OH) 2 D or the lack of production of 1,25(OH) 2 D. The first insight into the cause of this disorder was when 1,25(OH) 2 D 3 was chemically synthesized and provided to these patients. Within several months there was a dramatic improvement in their serum calcium and phosphorus level, with a decrease in alkaline phosphatase and PTH. Thus, it was concluded that vitamin D-dependent rickets was caused by a hereditary defect in the 1-OHase.
The cloning of the 1-OHase gene led to the identification of inactivating mutations that confirmed the hypothesis for the cause of this rare genetic disorder. Patients with PDDR present in their first year of life with severe hypocalcemia, which can cause seizures and carpal pedal spasms, hypophosphatemia, elevated alkaline phosphatase, and PTH. Their blood level of 25(OH)D is usually normal, and treating them with physiologic doses of vitamin D had little effect on correcting their abnormal biochemistries. The hallmark for making the diagnosis is a low or undetectable blood level of 1,25(OH) 2 D.
If these patients are not appropriately treated with replacement doses of 1,25(OH) 2 D 3 , they will have the same skeletal deformities seen in children with severe vitamin D deficiency ( Fig. 13.3 ). These include global poor mineralization of the skeleton and skeletal manifestations in areas of rapid bone growth, including the long bones, epiphyses, and costochondral junctions. Classic clinical manifestation of rickets is usually observed between the ages of 4–12 months. The first skeletal deformities that are observed are the classic rachitic rosary due to hypertrophy of the costochondral junctions leading to beading along both sides of the sternum. There is also involution of the ribs, protrusion of the sternum (pigeon chest), recession of the costochondral junctions, and traverse depressions causing Harrison’s groove. Chondrocyte hypertrophy is disrupted, and as a result, there is uncontrolled chondrocyte hypertrophy, leading to expansion of the epiphyseal plates, which appears as widening of the wrists and ends of the other long bones. Once the child begins to stand, gravity pushing on the lower limbs results in an inward (genu valgum) or outward (genu varum) bowing of the tibia and femur. Poor mineralization of the skull in the occipital region causes rachitic craniotabes, enlarged sutures and fontanelles, delayed closing of fontanelles, and occipital or parietal flattening, causing squaring of the cranium and frontal bossing. Tooth development and eruption is impaired and delayed, and the hypoplasia of the enamel leads to early dental caries.
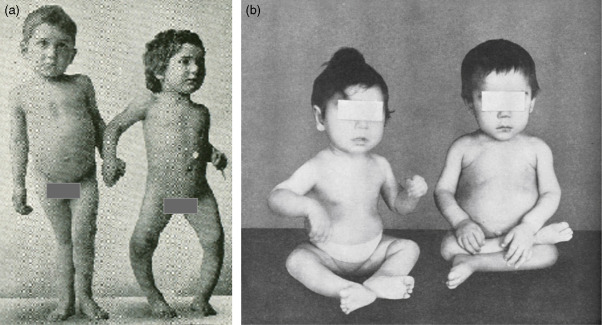
Extraskeletal manifestations associated with hypocalcemia lead to tetany, seizures, laryngospasm and hypocalcemic myocardiopathy, and death. These children have a delay in motor development, causing severe muscle weakness, especially of the proximal muscles and thoracic muscles. The combination of poor thoracic muscular function and softening of the rib cage leads to poor ventilation, increasing the risk of upper respiratory tract infections and respiratory obstruction.
These patients respond well to replacement doses of 1–2 μg of 1,25(OH) 2 D 3 (calcitriol) along with adequate calcium intake. The serum calcium levels begin to rise within 24 h, and radiologic healing is observed by 3 months.
Vitamin D-Dependent Rickets Type II: Hereditary Vitamin D Resistant Rickets (HVDRR)
Children who had severe biochemical abnormalities and skeletal abnormalities associated with vitamin D deficiency rickets and who did not respond to physiologic doses of vitamin D and only rarely responded to pharmacologic doses of vitamin D were considered to have vitamin D-dependent rickets. However, unlike children with PDDR who had a low or undetectable blood level of 1,25(OH) 2 D, these children had a markedly elevated blood level of 1,25(OH) 2 D. It was assumed that these children must have a genetic defect causing a lack of responsiveness to the calcium and bone metabolism effects of 1,25(OH) 2 D. The most likely cause was a defective or absent recognition of 1,25(OH) 2 D due to a mutation of the VDR. These patients had variable responses to replacement doses of 1,25(OH) 2 D 3 . There has been a multitude of point mutations of the VDR identified, causing disruption of hormone binding or DNA binding, leading to partial resistance to 1,25(OH) 2 D 3 replacement therapy. In some cases, these patients respond to pharmacologic doses of 1,25(OH) 2 D 3 . However, some patients have a point mutation that prevents the production of VDR or prevents the VDR from either binding to 1,25(OH) 2 D or permitting the VDR-RXR-1,25(OH) 2 D complex from binding to its responsive element within the DNA. These patients often are resistant to both physiologic and pharmacologic doses of 1,25(OH) 2 D 3 . (See Table 13.1 .)
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


